name: inverse layout: true class: center, middle, inverse <div class="my-header"><span> <a href="/training-material/topics/dev" title="Return to topic page" ><i class="fa fa-level-up" aria-hidden="true"></i></a> <a href="https://github.com/galaxyproject/training-material/edit/main/topics/dev/tutorials/conda/slides.html"><i class="fa fa-pencil" aria-hidden="true"></i></a> </span></div> <div class="my-footer"><span> <img src="/training-material/assets/images/GTN-60px.png" alt="Galaxy Training Network" style="height: 40px;"/> </span></div> --- <img src="/training-material/assets/images/GTNLogo1000.png" alt="Galaxy Training Network" class="cover-logo"/> <br/> <br/> # Tool Dependencies and Conda <br/> <br/> <div markdown="0"> <div class="contributors-line"> <ul class="text-list"> <li> <a href="/training-material/hall-of-fame/nsoranzo/" class="contributor-badge contributor-nsoranzo"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/nsoranzo?s=36" alt="Nicola Soranzo avatar" width="36" class="avatar" /> Nicola Soranzo</a> <li> <a href="/training-material/hall-of-fame/jmchilton/" class="contributor-badge contributor-jmchilton"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/jmchilton?s=36" alt="John Chilton avatar" width="36" class="avatar" /> John Chilton</a> <li> <a href="/training-material/hall-of-fame/bgruening/" class="contributor-badge contributor-bgruening"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/bgruening?s=36" alt="Björn Grüning avatar" width="36" class="avatar" /> Björn Grüning</a> <li> <a href="/training-material/hall-of-fame/hmenager/" class="contributor-badge contributor-hmenager"><img src="https://avatars.githubusercontent.com/hmenager?s=36" alt="Hervé Ménager avatar" width="36" class="avatar" /> Hervé Ménager</a></li> </ul> </div> </div> <!-- modified date --> <div class="footnote" style="bottom: 8em;"> <i class="far fa-calendar" aria-hidden="true"></i><span class="visually-hidden">last_modification</span> Updated: <i class="fas fa-fingerprint" aria-hidden="true"></i><span class="visually-hidden">purl</span><abbr title="Persistent URL">PURL</abbr>: <a href="https://gxy.io/GTN:S00045">gxy.io/GTN:S00045</a> </div> <!-- other slide formats (video and plain-text) --> <div class="footnote" style="bottom: 5em;"> <i class="fas fa-file-alt" aria-hidden="true"></i><span class="visually-hidden">text-document</span><a href="slides-plain.html"> Plain-text slides</a> | </div> <!-- usage tips --> <div class="footnote" style="bottom: 2em;"> <strong>Tip: </strong>press <kbd>P</kbd> to view the presenter notes | <i class="fa fa-arrows" aria-hidden="true"></i><span class="visually-hidden">arrow-keys</span> Use arrow keys to move between slides </div> ??? Presenter notes contain extra information which might be useful if you intend to use these slides for teaching. Press `P` again to switch presenter notes off Press `C` to create a new window where the same presentation will be displayed. This window is linked to the main window. Changing slides on one will cause the slide to change on the other. Useful when presenting. --- ## Requirements Before diving into this slide deck, we recommend you to have a look at: - [Development in Galaxy](/training-material/topics/dev) - Tool development and integration into Galaxy: [<i class="fab fa-slideshare" aria-hidden="true"></i><span class="visually-hidden">slides</span> slides](/training-material/topics/dev/tutorials/tool-integration/slides.html) - [<i class="fas fa-laptop" aria-hidden="true"></i><span class="visually-hidden">tutorial</span> hands-on](http://planemo.readthedocs.io/en/latest/writing_standalone.html) - Prerequisites for building software/conda packages: [<i class="fab fa-slideshare" aria-hidden="true"></i><span class="visually-hidden">slides</span> slides](/training-material/topics/dev/tutorials/conda_sys/slides.html) --- ### <i class="far fa-question-circle" aria-hidden="true"></i><span class="visually-hidden">question</span> Questions - How can I connect tools to applications and libraries? - What are the advantages of declaring dependencies for my tool? - What are Conda and Bioconda? - What are Conda recipes and environments? - How do I find and use existing Conda recipes? - How do I develop Conda recipes for use within Galaxy tools? --- ### <i class="fas fa-bullseye" aria-hidden="true"></i><span class="visually-hidden">objectives</span> Objectives - Learn to use existing Conda recipes to enable best practice tool dependency management in Galaxy. - Learn the basics of building Conda recipes and contributing to Bioconda. - Learn to use Planemo to assist in developing Galaxy tools from existing and new Conda recipes. --- ## Planemo These slides mirror the section on "Dependencies and Conda" in the [Planemo Documentation](https://planemo.readthedocs.io/en/latest/writing_advanced.html#dependencies-and-conda). --- ## Galaxy Dependencies --- class: left, enlarge120 ### Example Tool (1 / 2) From Planemo docs - the following example builds a tool for the `seqtk seq` command. ```sh $ planemo tool_init --force \ --id 'seqtk_seq' \ --name 'Convert to FASTA (seqtk)' \ --requirement seqtk@1.2 \ --example_command 'seqtk seq -a 2.fastq > 2.fasta' \ --example_input 2.fastq \ --example_output 2.fasta \ --test_case \ --cite_url 'https://github.com/lh3/seqtk' \ --help_from_command 'seqtk seq' ``` Notice the `--requirement seqtk@1.2`. --- class: left, enlarge120 ### Example Tool (2 / 2) The `--requirement seqtk@1.2` gets translated into the following Galaxy tool XML: ```xml <requirements> <requirement type="package" version="1.2">seqtk</requirement> </requirements> ``` --- ## Dependency Resolution 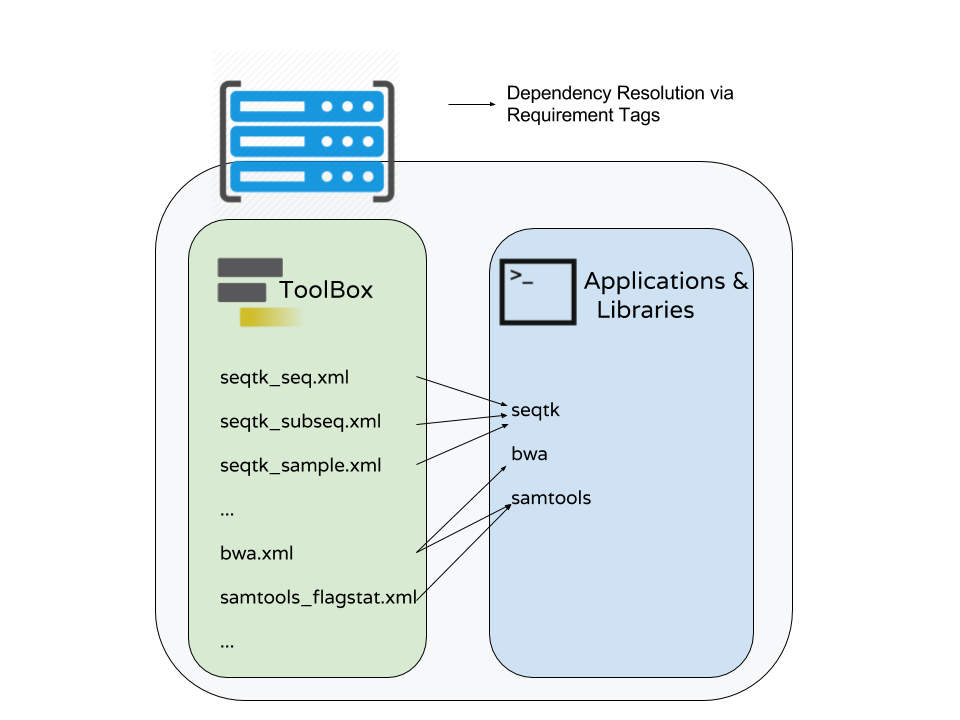 ??? - Notice that multiple tools may be mapped to the same requirements and any tools may use multiple Conda recipes. - There are few different ways to populate Applications and Libraries on the right - we will talk about Conda which is what we consider the "community best practice". --- class: enlarge120  Package, dependency and environment management --- class: enlarge120 ###.image-25[] <br/> Conda Terminology Conda **recipes** build **packages** that are published to **channels**. --- class: enlarge120 ### .image-25[] <br /> Conda Key Features for Galaxy - No compilation at install time - *binaries* with their dependencies, libraries... - Support for all operating systems Galaxy targets - Easy to manage *multiple versions* of the same recipe - HPC-ready: no root privileges needed - Easy-to-write YAML recipes - Vibrant Communities ??? - Recipes: independent of the programming language in which software is written - Support for multiple versions at the same time is needed for reproducibility Compared with the Tool Shed dependency management (`tool_dependencies.xml`), BioConda is: - More popular! - Easier to develop. - Easier to install and test. --- ###.image-25[] <br/> Conda Distributions ``` ``` 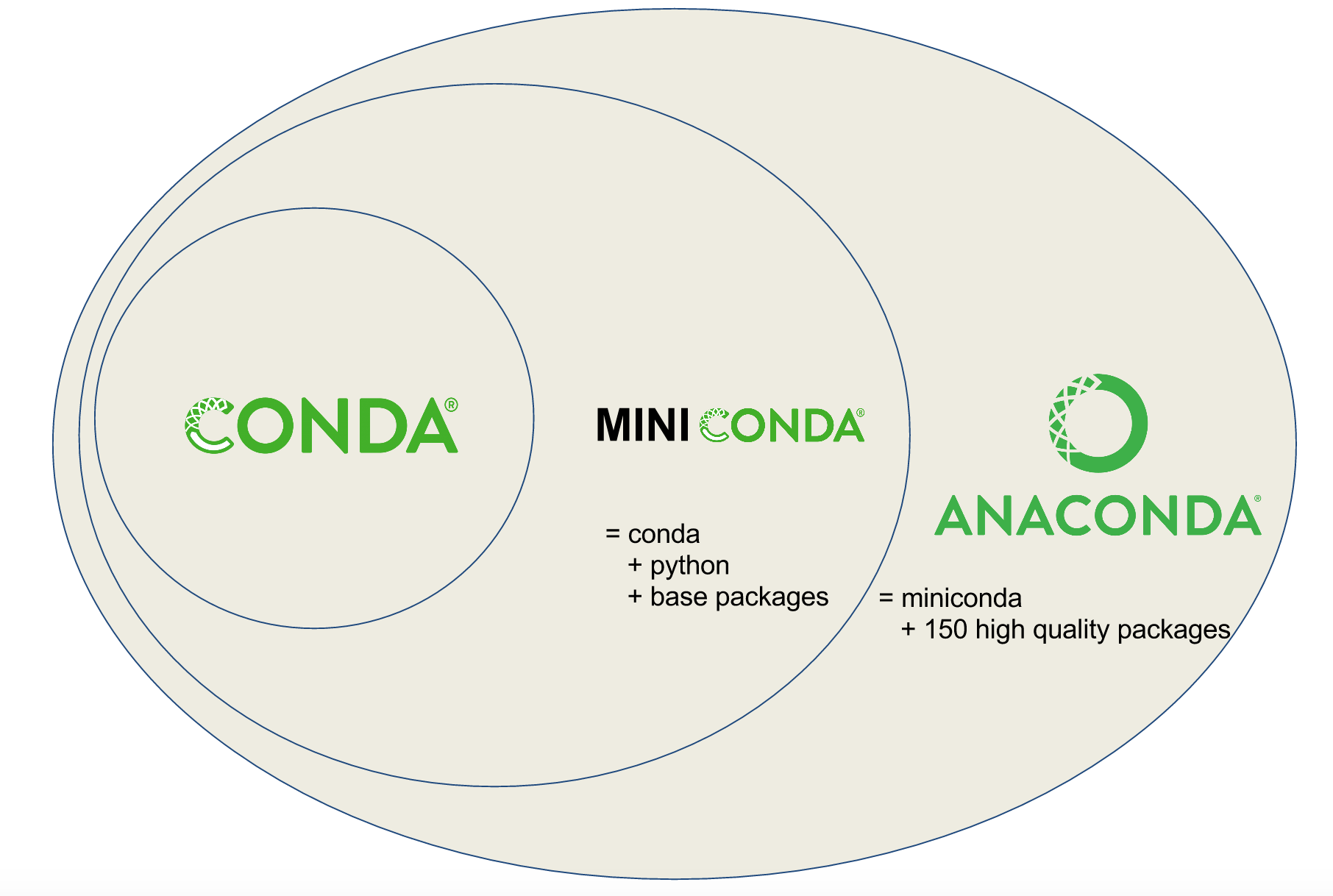 --- class: enlarge120 ###.image-25[] <br/> Best Practice Channels - Packages through channels within Continuum. - Conda channels searched by Galaxy for packages - iuc (legacy) - conda-forge - bioconda - defaults .footnote[If you are interested in Natural Language Processing or Cheminformatics you may be asking if these channels can still work for your tools. Despite the name [Bioconda](https://bioconda.github.io/) - it is really more about community and a set of best practices than about bioinformatics purity - many diverse packages have been integrated.] --- class: left, enlarge120 ###.image-25[] <br/> Install and Configure ```bash $ planemo conda_init $ export PATH=$PATH:~/miniconda3/bin $ which conda /Users/john/miniconda3/bin/conda ``` Planemo installs Conda using miniconda and configured defaults designed to easy development. This has already been done on [Planemo machine](https://github.com/galaxyproject/planemo-machine). --- class: enlarge120 ###.image-25[] <br/> Quickstart #### Using Conda outside Planemo - Install some packages within an isolated environment ```bash $ conda create -n yaml pyyaml $ conda env list base * ~/miniconda3 yaml ~/miniconda3/envs/yaml $ conda activate yaml (yaml) $ ``` - Install a package in the current environment ```bash $ conda install pyyaml ``` --- class: enlarge150 ## Conda and Galaxy Galaxy now automatically installs Conda when first launched and will use [Bioconda](https://bioconda.github.io/) and other channels for package resolution. --- ### Installing Tools with Conda 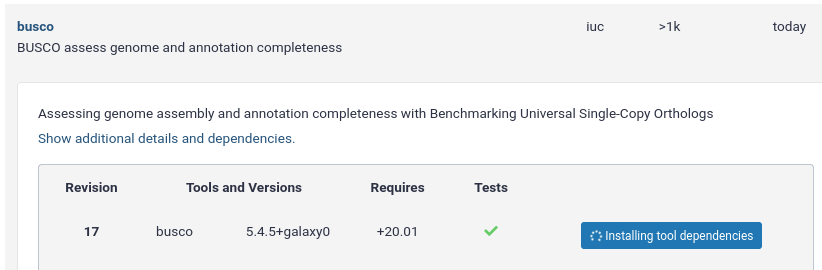 --- ### Managing Tool Dependencies 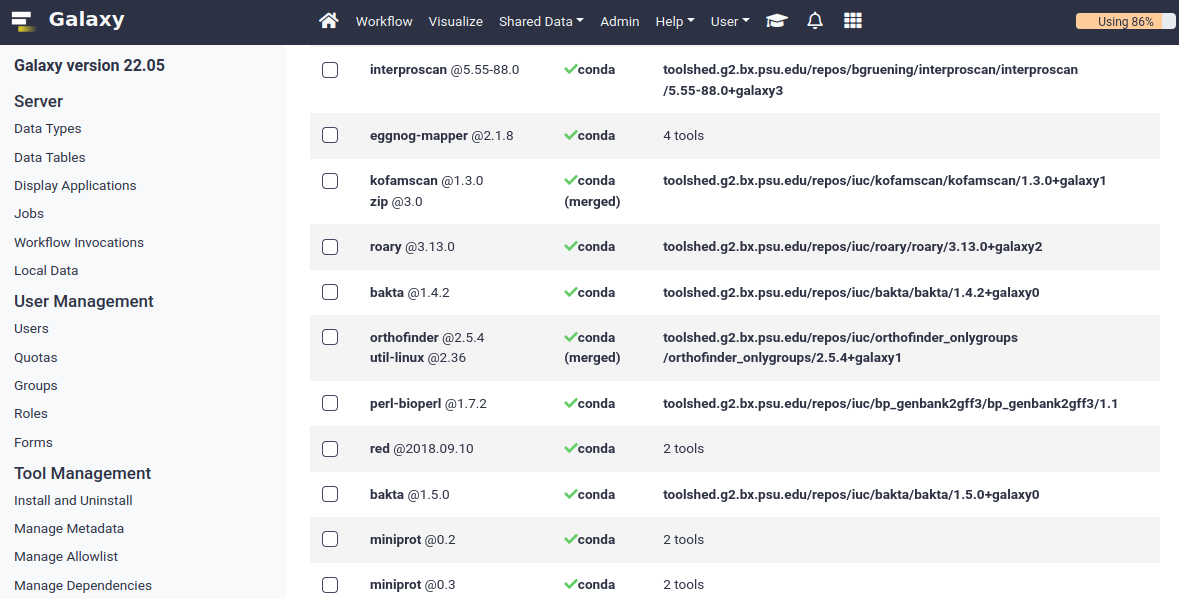 --- class: enlarge150 ## Conda and Planemo Using Conda directly is generally package-centric, Planemo provides abstractions that are tool-centric. --- class: left, enlarge120 The next few slides will use the seqtk example from Planemo's documentation - this can be downloaded to follow along using the following command: ```bash $ planemo project_init --template=seqtk_complete seqtk_example $ cd seqtk_example ``` --- class: left ### Linting Conda Dependencies .enlarge120[Planemo can check if the requirements of a tool are available in best practice Conda channels using the `--conda_requirements` flag of `planemo lint`.] <br> ```bash $ planemo lint --conda_requirements seqtk_seq.xml Linting tool /Users/john/workspace/planemo/docs/writing/seqtk_seq_v6.xml ... Applying linter requirements_in_conda... CHECK .. INFO: Requirement [seqtk@1.2] matches target in best practice Conda channel [bioconda]. ``` <br> .enlarge120[*Notice Planemo indicates this tool is available and shows the channel it is available in.*] --- class: left ### The Planemo `conda_install` command .reduce70[```sh $ planemo conda_install seqtk_seq.xml Install conda target CondaTarget[seqtk,version=1.2] /home/john/miniconda3/bin/conda create -y --name __seqtk@1.2 seqtk=1.2 Fetching package metadata ............... Solving package specifications: .......... Package plan for installation in environment /home/john/miniconda3/envs/__seqtk@1.2: The following packages will be downloaded: package | build ---------------------------|----------------- seqtk-1.2 | 0 29 KB bioconda The following NEW packages will be INSTALLED: seqtk: 1.2-0 bioconda zlib: 1.2.8-3 Fetching packages ... .... # # To deactivate this environment, use: # > source deactivate __seqtk@1.2 # $ which seqtk seqtk not found ```] Notice seqtk hasn't been placed on the `PATH`, an environment has been setup that Galaxy (when used through Planemo) can leverage. --- class: enlarge120 ### The Planemo `conda_env` command .reduce70[```sh $ . <(planemo conda_env seqtk_seq.xml) Deactivate environment with conda_env_deactivate (seqtk_seq) $ which seqtk /home/planemo/miniconda2/envs/jobdepsiJClEUfecc6d406196737781ff4456ec60975c137e04884e4f4b05dc68192f7cec4656/bin/seqtk (seqtk_seq) $ seqtk seq Usage: seqtk seq [options] <in.fq>|<in.fa> Options: -q INT mask bases with quality lower than INT [0] -X INT mask bases with quality higher than INT [255] -n CHAR masked bases converted to CHAR; 0 for lowercase [0] -l INT number of residues per line; 0 for 2^32-1 [0] ... -V shift quality by '(-Q) - 33' -U convert all bases to uppercases -S strip of white spaces in sequences (seqtk_seq) $ conda_env_deactivate $ ```] --- ## Using the Tool Environment Now that we have verified the Conda environment setup with `conda_install` works properly on the command-line, we can use our tool! `planemo test` and `planemo serve` will use this environment by default now for this tool. --- class: left ### Planemo `test` .reduce70[```sh $ planemo test seqtk_seq.xml ... INFO [galaxy.tools.actions] Handled output named output1 for tool seqtk_seq (20.136 ms) INFO [galaxy.tools.actions] Added output datasets to history (12.782 ms) INFO [galaxy.tools.actions] Verified access to datasets for Job[unflushed,tool_id=seqtk_seq] (10.954 ms) INFO [galaxy.tools.actions] Setup for job Job[unflushed,tool_id=seqtk_seq] complete, ready to flush (21.053 ms) INFO [galaxy.tools.actions] Flushed transaction for job Job[id=2,tool_id=seqtk_seq] (26.510 ms) INFO [galaxy.jobs.handler] (2) Job dispatched DEBUG [galaxy.tools.deps] Using dependency seqtk version 1.2 of type conda DEBUG [galaxy.tools.deps] Using dependency seqtk version 1.2 of type conda INFO [galaxy.jobs.command_factory] Built script [/tmp/tmpLvKwta/job_working_directory/000/2/tool_script.sh] for tool command [[ "$CONDA_DEFAULT_ENV" = "/Users/john/miniconda2/envs/__seqtk@1.2" ] || . /Users/john/miniconda2/bin/activate '/Users/john/miniconda2/envs/__seqtk@1.2' >conda_activate.log 2>&1 ; seqtk seq -a '/tmp/tmpLvKwta/files/000/dataset_1.dat' > '/tmp/tmpLvKwta/files/000/dataset_2.dat'] DEBUG [galaxy.tools.deps] Using dependency samtools version None of type conda DEBUG [galaxy.tools.deps] Using dependency samtools version None of type conda ok ---------------------------------------------------------------------- XML: /private/tmp/tmpLvKwta/xunit.xml ---------------------------------------------------------------------- Ran 1 test in 15.936s OK ```] .enlarge120[ The following line indicates the seqtk package was found: ``` [galaxy.tools.deps] Using dependency seqtk version 1.2 of type conda ``` ] --- <hands-on-title>Hands-on</hands-on-title>  --- <hands-on-title>Hands-on</hands-on-title> #### The Goal - Use the Planemo commands `conda_install`, `conda_env`, and `test` to practice the Galaxy tool dependency development lifecycle. --- <hands-on-title>Hands-on</hands-on-title> #### Steps Run the following commands to practice working with Galaxy tools, Planemo, and Conda. ```bash $ planemo project_init --template=seqtk_complete seqtk_example ``` ```bash $ cd seqtk_example ``` ```bash $ planemo conda_install seqtk_seq.xml ``` ```bash $ . <(planemo conda_env seqtk_seq.xml) ``` ```bash $ planemo test seqtk_seq.xml ``` --- class: enlarge120 ## Finding the Correct Requirements & Packages The previous example worked because a published Bioconda recipe named `seqtk` at version `1.2` was previously published, but how can these be found? Two easy approaches are using `planemo conda_search` and using the Anaconda web search. --- class: left ### Using the Planemo `conda_search` Command The Planemo `conda_search` command is a shortcut around `conda search` that searches best practice channels that Galaxy is configured to work with: ```sh $ planemo conda_search seqt Fetching package metadata ............... seqtk r75 0 bioconda r82 0 bioconda r93 0 bioconda 1.2 0 bioconda ``` Alternatively, `conda` can be used directly: ```sh $ $HOME/miniconda3/bin/conda search -c iuc -c conda-forge -c bioconda seqtk ``` --- ### Using Anaconda Search - https://anaconda.org  --- ### Using Anaconda Search - https://anaconda.org  ??? Notice that only one of these results is a best practice channel and so that is the only one that will be used by Galaxy by default. --- <hands-on-title>Hands-on</hands-on-title>  --- <hands-on-title>Hands-on</hands-on-title> #### The Goal - Find the correct package and version for a tool in a best practice channel. - Add a requirement to a tool to allow Galaxy to find, install, and use a Conda package. --- <hands-on-title>Hands-on</hands-on-title> #### Steps 1. Run the following commands to download an example tool to modify. ```sh $ planemo project_init --template conda_exercises conda_exercises $ cd conda_exercises/exercise1 $ ls pear.xml test-data ``` 2. Run `planemo test pear.xml` to verify the tool does not function without dependencies defined. 3. Use `--conda_requirements` flag with `planemo lint` to verify it does indeed lack requirements. 4. Use `planemo conda_search` or the [Anaconda](https://anaconda.org/) website to search for the correct package and version in a best practice channel. 5. Update pear.xml with the correct requirement tags. 6. Re-run the `lint` command from above to verify the tool now has the correct dependency definition. 7. Re-run the `test` command from above to verify the tool test now works properly. --- ## Writing a Conda recipe - If searching best practice channels fails, you may need to build a Conda recipe. - A Conda recipe is defined by a directory, the two most important files in this directory are: - `meta.yaml`: contains all the metadata of the recipe - `build.sh`: the (optional) Unix script that installs the files .footnote[[conda-build user guide](https://conda.io/projects/conda-build/en/latest/user-guide/index.html) <br/> [Bioconda guidelines](https://bioconda.github.io/contributor/index.html)] --- class: left ## `meta.yaml` `meta.yaml` contains basic metadata about the recipe. ```yaml {% set version = "0.7.17" %} {% set sha256 = "980b9591b61c60042c4a39b9e31ccaad8d17ff179d44d347997825da3fdf47fd" %} package: name: bwa version: {{ version }} source: url: https://github.com/lh3/bwa/archive/v{{ version }}.tar.gz sha256: {{ sha256 }} patches: - Makefile.patch build: number: 7 about: home: https://github.com/lh3/bwa license: GPL3 license_file: COPYING summary: The BWA read mapper. ``` --- ## `meta.yaml` > `requirements` ```yaml requirements: build: - {{ compiler('c') }} host: - zlib run: - zlib - perl ``` <small> - `build`: requirements needed to perform the compilation (if any) - `host`: requirements needed during the build step (here compilation) - `run`: requirements needed at runtime </small> --- ## Preprocessing Selectors - Common selectors include `linux`, `osx`, `py2k`, `py3k`. - Evaluated as Python expressions - feel free to use `and`, `or`, etc. ```yaml requirements: build: - bz2file # [py < 33] - typing # [py27 or py34] ``` ```yaml build: skip: True # [osx] ``` .footer[.center[https://conda.io/projects/conda-build/en/latest/source/define-metadata.html#preprocessing-selectors ]] --- class: left ## Tests `meta.yaml` should contain simple tests. These are commands executed at the end of `conda build` and expected to return `0` on success. ```yaml test: commands: - bowtie2 --version ``` ```yaml test: commands: - bwa 2>&1 | grep 'index sequences in the' ``` ```yaml test: commands: - '$R -e "library(''xcms'')"' ``` Please note that the Conda tests run inside the runtime environment and not in the build environment. .footnote[.center[https://conda.io/projects/conda-build/en/latest/source/define-metadata.html#test-section ]] --- From the [Bioconda Guidelines](https://bioconda.github.io/contributor/guidelines.html): > An adequate test must be included in the recipe. An "adequate" test depends on the recipe, but must be able to detect a successful installation. While many packages may ship their own test suite (unit tests or otherwise), including these in the recipe is not recommended since it may timeout the build system on CircleCI. We especially want to avoid including any kind of test data in the repository. --- ## `build.sh` ```bash #!/bin/bash ./configure --prefix=$PREFIX make make install ``` ```bash #!/bin/bash mkdir -p $PREFIX/bin cp *.py $PREFIX/bin ``` <small> - `$PREFIX` is a variable defined by conda when building the package. - `$PREFIX` usually contains `bin/`, `lib/`, and `include/` that are filled with the package files. - `$PREFIX` will be loaded in the user environment at runtime: `PATH`, `LD_LIBRARY_PATH`, `PYTHONPATH`, ... </small> .footnote[[http://training.galaxyproject.org/training-material/topics/dev/tutorials/conda_sys/slides.html](/training-material/topics/dev/tutorials/conda_sys/slides.html)] --- ## Skeletons ```bash $ conda skeleton pypi <packagename> ``` ```bash $ conda skeleton cran <packagename> ``` ```bash $ bioconductor_skeleton.py <packagename> ``` ```bash $ conda skeleton cpan <packagename>` ``` These generate pre-filled recipes <small>(not guaranteed to work out of the box)</small> for specific programming environments. .footnote[[Building conda packages with conda skeleton](https://docs.conda.io/projects/conda-build/en/latest/user-guide/tutorials/build-pkgs-skeleton.html)] --- ## Building Once the recipe is ready to go, the `conda build` command can be used to build it. ```bash $ $HOME/miniconda3/bin/conda build . ``` 1. BUILD START: Builds/Compiles the package 2. BUILD START: Provides a .tar.bz2 3. TEST START: Installs the .tar.bz2 previously generated 4. TEST START: Launches the functional tests 5. (Provides the .tar.bz2 path) .footnote[If miniconda wasn't configured with `planemo conda_init`, you may have <br/> to run `conda install conda-build` before using the above command.] --- .image-75[] - A channel dedicated to bioinformatics (and other informatics) packages - https://bioconda.github.io - https://anaconda.org/bioconda - Open to [contribution](https://bioconda.github.io/contributing.html) - GitHub repository: https://github.com/bioconda/bioconda-recipes --- ###.image-25[] contributing 1/2 - Fork [Bioconda](https://github.com/bioconda/bioconda-recipes/fork). - Clone your fork: ```bash $ git clone https://github.com/ <myuser> /bioconda-recipes ``` - Create a new branch `package` ```bash $ git checkout -b package ``` - Fill two files `meta.yaml` and `build.sh` in a new recipe directory - Build your new package and test it using `conda build` --- ###.image-25[] contributing 2/2 - Commit and push to GitHub ```bash $ git add recipe ``` ```bash $ git commit -m 'my recipe description' ``` ```bash $ git push origin package ``` - Create a [Pull Request](https://github.com/bioconda/bioconda-recipes) - After the PR is merged, wait for the functional tests to pass on the master branch - Enjoy your new Conda package at https://anaconda.org/bioconda See [Contributing with GitHub](/training-material/topics/contributing/tutorials/github-command-line-contribution/slides.html). --- ### Planemo and `--conda_use_local` By default, Galaxy and Planemo will ignore locally built packages. Simply pass `--conda_use_local` to various Planemo commands (e.g. `test`, `conda_install`, or `serve`) to use the local package cache. *Enables developing Galaxy tools and Conda recipes in parallel.* --- <hands-on-title>Hands-on</hands-on-title>  --- <hands-on-title>Hands-on</hands-on-title> #### The Goal - Implement and test a local Conda recipe. - Use Planemo and Galaxy with a locally built package. --- class: left <hands-on-title>Hands-on</hands-on-title> #### Before If you have completed `exercise1`, open `exercise2`. ```sh $ cd ../exercise2 $ ls fleeqtk_seq.xml test-data ``` This directory contains the outline of a tool for [fleeqtk](https://github.com/jmchilton/fleeqtk). fleeqtk is a fork of the project seqtk that many Planemo tutorials are built around and the example tool `fleeqtk_seq.xml` should be fairly familiar. --- class: left <hands-on-title>Hands-on</hands-on-title> #### Steps 1. Clone and branch Bioconda (https://github.com/bioconda/bioconda-recipes) 2. Build a recipe for fleeqtk version 1.3. You may wish to use conda skeleton, start from scratch, or copy the recipe of seqtk and work from there - any of these strategies should work * fleeqtk 1.3 can be downloaded using the URL https://github.com/jmchilton/fleeqtk/archive/v1.3.tar.gz * fleeqtk can be built using `make` and installed with `make install` 3. Use `conda build` to build the recipe 4. Add a `requirement` for this new package in the example tool. 5. Run `planemo conda_install --conda_use_local fleeqtk_seq.xml` to install the package for Galaxy 6. Run `planemo test fleeqtk_seq.xml` to verify the tool and package work together --- # Advanced Topics in Conda Development --- ## Jinja Templating ```yaml {% set name = "seqtk" %} {% set version = "1.15.1" %} package: name: {{ name }} version: {{ version }} source: url: http://coolsoftware.com/{{ name }}/{{ version }}/{{ name }}-{{ version }}.zip ``` https://conda.io/projects/conda-build/en/latest/source/define-metadata.html#templating-with-jinja --- class: left ## Stable URLs ```yaml source: url: https://github.com/lh3/bwa/archive/v0.7.15.tar.gz md5: 54fdee953c5c256d36885a1c5c6b118c ``` > While supported by Conda, `git_url` and `git_rev` are not as stable as a git tarball. Ideally a github repo should have tagged releases that are accessible as tarballs from the “releases” section of the github repo. In addition tarballs can be easily mirrored and Bioconda is saving a copy of every tarball so the recipe can be rebuild at any time. --- class: left ## Python For PyPI packages ```sh conda skeleton pypi <package_name> ``` - Builds likely correct `build.sh` and `meta.yaml`. - The test automatically added is probably sufficient for library, may need to write extra tests for command-line tools. - Recipes requiring `python` should build on Python 2.7, 3.5, and 3.6 by default, preprocessing selectors can be used with `build: skip` to skip targets. --- ## Python - pysam's `build.sh` ```sh #!/bin/bash # Remove gcc statements that do not work on older compilers for CentOS5 # support sed -i'' -e 's/"-Wno-error=declaration-after-statement",//g' setup.py sed -i'' -e 's/"-Wno-error=declaration-after-statement"//g' setup.py # linking htslib, see: # https://pysam.readthedocs.org/en/latest/installation.html#external # https://github.com/pysam-developers/pysam/blob/v0.9.0/setup.py#L79 export CFLAGS="-I$PREFIX/include" export CPPFLAGS="-I$PREFIX/include" export LDFLAGS="-L$PREFIX/lib" export HTSLIB_LIBRARY_DIR=$PREFIX/lib export HTSLIB_INCLUDE_DIR=$PREFIX/include $PYTHON setup.py install ``` --- ## Python - pysam's `meta.yaml` .reduce50[ ```yaml {% set version = "0.15.2" %} {% set samtools_version = "1.9" %} {% set bcftools_version = "1.9" %} package: name: pysam version: '{{ version }}' source: url: https://github.com/pysam-developers/pysam/archive/v{{ version }}.tar.gz sha256: 8cb3dd70f0d825086ac059ec2445ebd2ec5f14af73e7f1f4bd358966aaee5ed3 build: number: 3 binary_relocation: False # [linux] requirements: build: - {{ compiler('c') }} host: - htslib - samtools {{ samtools_version }} - bcftools {{ bcftools_version }} - cython - python - setuptools - zlib - curl - libdeflate run: - samtools {{ samtools_version }} - bcftools {{ bcftools_version }} - python - curl - libdeflate test: imports: - pysam ```] --- ## R For CRAN packages ```sh conda skeleton cran <packagename> ``` - Builds likely correct `build.sh` and `meta.yaml`. - The recipe name will have an `r-` prefix and will be converted to lowercase. <i class="fas fa-exclamation-triangle" aria-hidden="true"></i><span class="visually-hidden">warning</span> _The majority of R packages on CRAN are generic and should therefore be submitted at Conda-Forge. Exceptions are r-\* packages that depends on bioconductor-\* packages._ [Conda-Forge contribution guidelines](https://conda-forge.org/docs/maintainer/adding_pkgs.html) --- ## Java - Recipes should use the `openjdk` package from [conda-forge](https://github.com/conda-forge/openjdk-feedstock). - Add a wrapper script if the software is typically called via `java -jar`. - JAR files should go in `$PREFIX/share/$PKG_NAME-$PKG_VERSION-$PKG_BUILDNUM` - A wrapper script should be placed here as well, and symlinked to $PREFIX/bin. https://bioconda.github.io/guidelines.html#java --- ## Java - PeptideShaker's `build.sh` ```sh #!/bin/bash set -eu -o pipefail outdir=$PREFIX/share/$PKG_NAME-$PKG_VERSION-$PKG_BUILDNUM mkdir -p $outdir mkdir -p $PREFIX/bin cp -R * $outdir/ cp $RECIPE_DIR/peptide-shaker.py $outdir/peptide-shaker ls -l $outdir ln -s $outdir/peptide-shaker $PREFIX/bin chmod 0755 "${PREFIX}/bin/peptide-shaker" ``` --- class: reduce70 ## Java - PeptideShaker's `meta.yaml` ```yaml ... source: url: http://genesis.ugent.be/maven2/eu/isas/peptideshaker/{{ name }}/{{ version }}/{{ name }}-{{ version }}.zip md5: 14a48413e28a25614f5fda2b381d7197 requirements: build: run: - openjdk >=6 - python test: commands: - peptide-shaker eu.isas.peptideshaker.cmd.PeptideShakerCLI - peptide-shaker eu.isas.peptideshaker.cmd.PeptideShakerCLI -Xms512m -Xmx1g ``` --- ## Perl For CPAN packages ```sh conda skeleton cpan <packagename> ``` - Builds likely correct `build.sh` and `meta.yaml`. - The recipe will have the `perl-` prefix. --- class: reduce70 ## Perl - Module-Build ```yaml package: name: perl-module-build version: "0.4214" source: url: https://cpan.metacpan.org/authors/id/L/LE/LEONT/Module-Build-0.4214.tar.gz md5: 7b7ca5a47bef48c50c8b5906ca3ac7fb build: number: 0 requirements: host: - perl - perl-cpan-meta-yaml - perl-extutils-parsexs - perl-data-dumper # [...] run: - perl - perl-text-parsewords - perl-cpan-meta - perl-version # [...] test: # Perl 'use' tests imports: - Module::Build - Module::Build::Base - Module::Build::Compat - Module::Build::Config # [...] about: home: https://metacpan.org/pod/Module::Build license: perl_5 summary: 'Build and install Perl modules ``` --- class: left ## Metapackages > Metapackages tie together other packages. All they do is define dependencies. For example, the hubward-all metapackage specifies the various other conda packages needed to get full hubward installation running just by installing one package. > Other metapackages might tie together conda packages with a theme. For example, all UCSC utilities related to bigBed files, or a set of packages useful for variant calling. https://bioconda.github.io/guidelines.html#metapackages --- class: left ## CircleCI Continuous Building .image-70[] - Lint recipes. - Build and run tests. - Build and publish Docker container. - Publish to anaconda.org. --- ## CircleCI Command Line Interface (CLI) - Installation: .reduce70[```bash $ curl -o /usr/local/bin/circleci https://circle-downloads.s3.amazonaws.com/releases/build_agent_wrapper/circleci ```] .reduce70[```bash $ chmod +x /usr/local/bin/circleci ```] - The extended building and testing done by CircleCI can be executed locally using the CircleCI CLI in the root directory of Bioconda - It should be run from the top-level dir of the repo. - Build and test recipes: .reduce70[```bash $ circleci build ```] --- ### <i class="fas fa-key" aria-hidden="true"></i><span class="visually-hidden">keypoints</span> Key points - Conda and Bioconda are Galaxy best practices for connecting Galaxy tools to underlying applications and libraries. - Leveraging Conda allows easy installation of your tool's dependencies by Galaxy deployers. - The Planemo commands `conda_search`, `conda_init`, `conda_install`, `lint`, `test`, and `serve` make it easy to search and use existing Conda recipes when developing tools. - Conda recipe skeletons, `conda build`, and with `planemo conda_install --conda_use_local` allow easy development of new Conda recipes at the same time as Galaxy tools that wrap them. - Bioconda is a Galaxy best practice Conda channel for recipe publication. - Bioconda has easy to follow contribution guidelines and is very welcoming to new contributors. --- ## Thank You! This material is the result of a collaborative work. Thanks to the [Galaxy Training Network](https://training.galaxyproject.org) and all the contributors! <div markdown="0"> <div class="contributors-line"> <table class="contributions"> <tr> <td><abbr title="These people wrote the bulk of the tutorial, they may have done the analysis, built the workflow, and wrote the text themselves.">Author(s)</abbr></td> <td> <a href="/training-material/hall-of-fame/nsoranzo/" class="contributor-badge contributor-nsoranzo"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/nsoranzo?s=36" alt="Nicola Soranzo avatar" width="36" class="avatar" /> Nicola Soranzo</a><a href="/training-material/hall-of-fame/jmchilton/" class="contributor-badge contributor-jmchilton"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/jmchilton?s=36" alt="John Chilton avatar" width="36" class="avatar" /> John Chilton</a><a href="/training-material/hall-of-fame/bgruening/" class="contributor-badge contributor-bgruening"><img src="/training-material/assets/images/orcid.png" alt="orcid logo" width="36" height="36"/><img src="https://avatars.githubusercontent.com/bgruening?s=36" alt="Björn Grüning avatar" width="36" class="avatar" /> Björn Grüning</a><a href="/training-material/hall-of-fame/hmenager/" class="contributor-badge contributor-hmenager"><img src="https://avatars.githubusercontent.com/hmenager?s=36" alt="Hervé Ménager avatar" width="36" class="avatar" /> Hervé Ménager</a> </td> </tr> <tr class="reviewers"> <td><abbr title="These people reviewed this material for accuracy and correctness">Reviewers</abbr></td> <td> <a href="/training-material/hall-of-fame/bgruening/" class="contributor-badge contributor-badge-small contributor-bgruening"><img src="https://avatars.githubusercontent.com/bgruening?s=36" alt="Björn Grüning avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/hexylena/" class="contributor-badge contributor-badge-small contributor-hexylena"><img src="https://avatars.githubusercontent.com/hexylena?s=36" alt="Helena Rasche avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/nsoranzo/" class="contributor-badge contributor-badge-small contributor-nsoranzo"><img src="https://avatars.githubusercontent.com/nsoranzo?s=36" alt="Nicola Soranzo avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/shiltemann/" class="contributor-badge contributor-badge-small contributor-shiltemann"><img src="https://avatars.githubusercontent.com/shiltemann?s=36" alt="Saskia Hiltemann avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/bebatut/" class="contributor-badge contributor-badge-small contributor-bebatut"><img src="https://avatars.githubusercontent.com/bebatut?s=36" alt="Bérénice Batut avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/abretaud/" class="contributor-badge contributor-badge-small contributor-abretaud"><img src="https://avatars.githubusercontent.com/abretaud?s=36" alt="Anthony Bretaudeau avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/lecorguille/" class="contributor-badge contributor-badge-small contributor-lecorguille"><img src="https://avatars.githubusercontent.com/lecorguille?s=36" alt="Gildas Le Corguillé avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/gallardoalba/" class="contributor-badge contributor-badge-small contributor-gallardoalba"><img src="https://avatars.githubusercontent.com/gallardoalba?s=36" alt="Cristóbal Gallardo avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/davebx/" class="contributor-badge contributor-badge-small contributor-davebx"><img src="https://avatars.githubusercontent.com/davebx?s=36" alt="Dave B. avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/astrovsky01/" class="contributor-badge contributor-badge-small contributor-astrovsky01"><img src="https://avatars.githubusercontent.com/astrovsky01?s=36" alt="Alex Ostrovsky avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/jmchilton/" class="contributor-badge contributor-badge-small contributor-jmchilton"><img src="https://avatars.githubusercontent.com/jmchilton?s=36" alt="John Chilton avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/willdurand/" class="contributor-badge contributor-badge-small contributor-willdurand"><img src="https://avatars.githubusercontent.com/willdurand?s=36" alt="William Durand avatar" width="36" class="avatar" /></a><a href="/training-material/hall-of-fame/njall/" class="contributor-badge contributor-badge-small contributor-njall"><img src="https://avatars.githubusercontent.com/njall?s=36" alt="Niall Beard avatar" width="36" class="avatar" /></a></td> </tr> </table> </div> </div> <div style="display: flex;flex-direction: row;align-items: center;justify-content: center;"> <img src="/training-material/assets/images/GTNLogo1000.png" alt="Galaxy Training Network" style="height: 100px;"/> </div> Tutorial Content is licensed under <a rel="license" href="http://creativecommons.org/licenses/by/4.0/">Creative Commons Attribution 4.0 International License</a>.<br/>