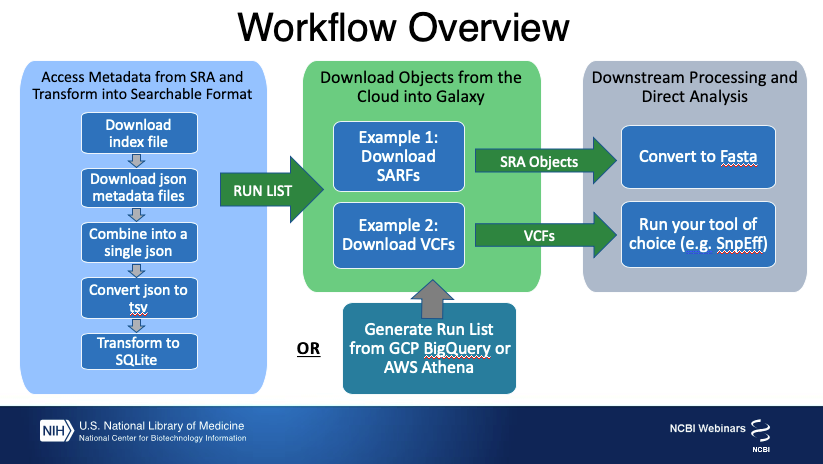
Traditionally, after a list of run accessions has been filtered on the NCBI website, the accessions are used to download and extract fastq using the SRA toolkit to enter into the next steps of the workflow. A newer compressed data type, generated from raw submitted data containing SARS-CoV-2 sequence, is also accessible to Galaxy users from SRA in the Cloud.
SRA Aligned Read Format (SARF) provides further output options beyond basic fastq format, for example:
contigs created from the raw reads in the run
reads aligned back to the contigs
reads with placeholder quality scores
VCF files can also be downloaded for these records relative to the SARS-CoV-2 RefSeq record
These formats can speed up workflows such as assembly and variant calling.
This data format is still referenced by the Run accession and accessed using the SRA toolkit.
This workshop describes the SARF data objects along with associated searchable metadata, and demonstrates a few ways to enter them into traditional workflows.
The aim of this tutorial is to introduce you to some of SRA’s new SARS-CoV-2 cloud resources and data formats, then show you how to filter for Runs of interest to you and access that data in your format of choice in Galaxy to use in your analysis pipeline.
SRA Aligned Read Format
All data submitted to SRA is scanned with our SARS-CoV-2 Detection Tool which uses a Kmer-based approach to identify Runs with Coronaviridae content. The initial scope of the project is limited to those runs deposited in SRA with at least 100 hits for SARS-CoV-2 via the SARS-CoV-2 Detection Tool, a read length of at least 75, and generated using the Illumina platform.
For these Runs, Saute was used to assemble contigs via guided assembly, with the SARS-CoV-2 refseq genomic sequence (NC_045512.2) used as the guide.
If contigs were successfully assembled, reads were mapped back to the contigs and coverage calculated. These contigs with the reads mapped back and with quality scores removed (to keep the object size small) are the aligned read format files.
The SRA toolkit can be used to dump just the contigs in fasta format, the reads aligned to the contigs in sam format or the raw reads in fastq format with placeholder quality scores.
The contigs were also assessed via megablast against the nucleotide blast database and the results made available for search.
In addition, to support investigation of viral evolution during the pandemic and after the introduction of vaccines, variants are identified relative to the SARS-CoV-2 RefSeq record for each processed run using BCFTools.
The SRA aligned reads, the VCF files, the results of these analyses (such as BLAST and VIGOR3 annotation), and the associated BioSample and sequencing library metadata are available for free access from cloud providers.
Comment
These data can be dumped in sam format using the sam-dump tool in the SRA Toolkit, but this function doesn’t work within Galaxy yet.
We hope to include that functionality in a future update.
Metadata for SARS-CoV-2 submissions to the SRA includes submitted sample and library information, BLAST results, descriptive contig statistics, and variation and annotation information. These metadata are updated daily and made available to query in the cloud using Google’s BigQuery or Amazon’s Athena services. However, the raw underlying information is also provided as a group of json files that can be downloaded for free from the Open Data Platform without logging in to the cloud. These json files can be imported to Galaxy and queried there to find Runs of interest.
Comment
Some of these tables include complex data fields (array of values) that don’t have a clean analogue in a classic SQL database or table and these can’t be easily queried in Galaxy currently. If you require access to cloud tables or fields not available in Galaxy we recommend accessing those natively in BigQuery or Athena.
We will import the JSON files into Galaxy to query them directory, however the files are split up for efficient querying in the cloud and updated daily, so we first need to get the most up-to-date list of files so we can import those to Galaxy. We’ll just be using a couple of tables in this training, but the other tables can be imported in the same way, using the index files below.
Comment
These metadata files are updated daily around 5:30pm EST. If you try to access the data around this time but encounter an error, trying again a short while later should resolve the issue. Time Zone Converter
Hands-on: Loading SRA Aligned Read Format (SARF) Object Metadata URLs into Galaxy
This step needs to be repeated at the beginning of an analysis to refresh the metadata to the latest daily version.
To create a new history simply click the new-history icon at the top of the history panel:
Rename your history, e.g. “NCBI SARF”
Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
Type the new name
Click on Save
To cancel renaming, click the galaxy-undo “Cancel” button
If you do not have the galaxy-pencil (Edit) next to the history name (which can be the case if you are using an older version of Galaxy) do the following:
Click on Unnamed history (or the current name of the history) (Click to rename history) at the top of your history panel
Type the new name
Press Enter
Click the upload icon toward the top left corner.
By default the familiar simple upload dialog should appear. This dialog has more advanced options as different tabs across the top of this dialog though.
From Rules menu select Add / Modify Column Definitions
Click Add Definition button and select URL
“URL”: A
Click Add Definition button and select Name (not name tag!)
“Name”: B
ClickApply. You should see a table with two columns, the left being the URL column, and the right being the Name column with just the filename.
You are now ready to start the upload.
Click the Upload button
With that you should have 7 different files in your history. If you examine the files you’ll see that they each have a list of filenames like the following table, with either today or yesterday’s date in the filename, depending on your timezone offset from NCBI’s offices. [Time zone converter](
From Rules menu, select Add / Modify Column Definitions
Add Definition, URL, Select Column C
Add Definition, List Identifier(s), Column A
Apply
Name the collection contigs.json
Click the Upload button.
Once those download jobs have all turned green in the history list, we’ll concatenate these into a single file.
Concatenate datasets ( Galaxy version 0.1.0) tail to head (cat) files with the following parameters:
param-collection“Datasets to Concatenate”: contigs.json collection
Click Run Tool
Rename this history item to contigs.single.json
Click on the galaxy-pencilpencil icon for the dataset to edit its attributes
In the central panel, change the Name field to contigs.single.json
Click the Save button
JQ ( Galaxy version 1.0) process JSON files with the following parameters:
param-file“JSON Input”: contigs.single.json
“jq filter”: [.[]]
“Convert output to tabular”: yes
Click Run Tool
Rename this file contigs.tsv
Click on the galaxy-pencilpencil icon for the dataset to edit its attributes
In the central panel, change the Name field to contigs.tsv
Click the Save button
Comment
The conversion of the json files to tabular format takes some time, this is a good time to go make some tea.
Query SARF Metadata
Now that a table has been generated, we will query the table to find the runs of interest. It is a good idea to save the table for future queries on the same dataset. Rerunning the import steps above without filtration will provide a different set of metadata each day.
Hands-on: Query the SRA Metadata Table using SQLite
Next we’ll query this metadata using the Query tabular tool to get a list of all Runs containing contigs of greater than 20,000 nucleotides and average coverage of at least 100X.
Run Query Tabular using sqlite sql ( Galaxy version 3.0.0) with the following parameters:
If you’re not using the GTN-in-Galaxy view, you can search for ‘sql’ to find it.
In “Database Table”:
param-repeat“Insert Database Table”
param-file“Tabular Dataset for Table”: contigs.tsv
If you plan to do multiple queries on the same SQL database or want to skip preprocessing the metadata for future work, it may be useful to set
param-repeat“Save the sqlite database in your history” to Yes
Click Run Tool and rename the output file to Run_list
Click on the galaxy-pencilpencil icon for the dataset to edit its attributes
In the central panel, change the Name field to Run_list
Click the Save button
Use Run_list to bring in Fastq files with Submitted Quality Scores (+BQS format)
We are not going to bring in the other metadata tables in this tutorial. Here is a list of column headers for contigs and the other tables.
You can find full definitions for these columns here:
If you would like to dump the raw, underlying data in fastq format with the original quality scores, you can stop here and use the Faster Download and Extract Reads in FASTQ ( Galaxy version 2.11.0+galaxy0) tool with the following parameters:
“input type”: list of SRA accessions, one per line
param-file“sra accession list”: Run_list
If you opted to conduct your metadata search in the cloud using AWS Athena or GCP BigQuery instead of importing the json file to Galaxy, you can save a list of your Run accessions from that search result and import that file as the Run_list to proceed with the rest of this tutorial.
Importing SARFs of Interest
Now that we have assembled a list of Runs that have contigs we are interested in, we’ll construct the path to the SARFS in the cloud and import those to Galaxy so we can work with them.
Hands-on: Importing SARFs of Interest
Upload Data
Open the Rule-based upload tab again, but this time:
“Upload data as”: Collection(s)
“Load tabular data from”: History Dataset
“Select dataset to load”: Run_list
ClickBuild to bring up the rule builder.
Make the following changes in the Rule Builder
From Column menu select Using a Regular Expression
From Rules menu select Add / Modify Column Definitions
Click Add Definition button and select URL
“URL”: B
Click Add Definition button and select List Identifier
“List Identifier”: A
Apply
Name the output collection sarf_path before clicking Upload
Comment
Please note that there can be some lag in availability of SARF/VCF files in the cloud (particularly for newly submitted data). So it’s possible to get a download error for a file that isn’t yet present in the cloud. In these cases waiting ~24 hours will generally resolve the issue and allow you to access the file.
Now we will use the SRA toolkit to retrieve the contigs in fasta format
Run Download and Extract Reads in FASTA/Q format from NCBI SRA ( Galaxy version 2.11.0+galaxy0) with the following parameters:
“Select input type”: SRA Archive in current history
param-collection“sra archive”: sarf_path
In “Advanced Options”:
“Table name within cSRA object”: REFERENCE
Click Run Tool
The resulting dataset includes the contigs generated from these runs with placeholder ? for quality scores
Rename this collection to sarf_contigs
Run Fastq to Fasta converter ( Galaxy version 1.1.5)
param-collection“FASTQ file to convert”: sarf_contigs (note: in the video this did not get renamed)
Click Run Tool
The resulting dataset includes the contigs generated from these Runs in fasta format
If you prefer to dump the raw reads in fastq format with placeholder quality scores, leave the Table name within cSRA object field blank.
Importing VCFs for SARS-Cov-2 Runs
This example starts with the same Run_list generated for importing SARFs.
A Run_list could also be imported after querying metadata in the cloud using Google’s BigQuery or Amazon’s Athena services. Metadata about these runs includes submitted sample and library information, BLAST results, descriptive contig statistics, and variation and annotations. See the tutorial video for a short demo on how to search and download Run_list from the cloud.
Hands-on: Importing VCFs of Interest
Upload Data
Open the Rule-based upload tab again, but this time:
“Upload data as”: Collection(s)
“Load tabular data from”: History Dataset
“Select dataset to load”: Run_list
ClickBuild to bring up the rule builder.
Make the following changes in the Rule Builder
From Column menu select Using a Regular Expression
From Rules menu select Add / Modify Column Definitions
“URL”: B
“List Identifier”: A
Apply
Name the output collection VCFs before clicking Upload
Comment
Please note that there can be some lag in availability of SARF/VCF files in the cloud (particularly for newly submitted data). So it’s possible to get a download error for a file that isn’t yet present in the cloud. In these cases waiting ~24 hours will generally resolve the issue and allow you to access the file.
Use VCFs in Another Galaxy Tool. Once you have imported the VCF files, you can use them in your standard pipeline- here we will annotate them with SnpEff.
Run SnpEff eff: annotate variants for SARS-CoV-2 ( Galaxy version 4.5covid19) with the following parameters:
param-collection“Sequence changes (SNPs, MNPs, InDels)”: the VCFs collection we just created
Comment
Please note that there are 2 Snepff tools, please choose the one for SARS-CoV-2
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{galaxy-interface-ncbi-sarf,
author = "Jon Trow and Adelaide Rhodes",
title = "SRA Aligned Read Format to Speed Up SARS-CoV-2 data Analysis (Galaxy Training Materials)",
year = "",
month = "",
day = "",
url = "\url{https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/ncbi-sarf/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol}
}
Congratulations on successfully completing this tutorial!
You can use Ephemeris's shed-tools install command to install the tools used in this tutorial.
Questions:
 Open image in new tab
Open image in new tabOpen image in new tab
