Small Non-coding RNA Clustering using BlockClust
| Author(s) |
|
| Reviewers |
|
OverviewQuestions:
Objectives:
What do the read profiles of small non-coding RNAs represent?
How to cluster the read profiles based on some intrinsic features?
Requirements:
Difference between mRNA-seq and smallRNA-seq
Unsupervised grouping of the adjacent reads into read profiles
Look and learn what the shapes of the read profiles represent
Clustering of the read profiles by machine learning algorithm
- Introduction to Galaxy Analyses
- slides Slides: Quality Control
- tutorial Hands-on: Quality Control
- slides Slides: Mapping
- tutorial Hands-on: Mapping
Time estimation: 1 hourSupporting Materials:Published: Dec 14, 2018Last modification: Nov 9, 2023License: Tutorial Content is licensed under Creative Commons Attribution 4.0 International License. The GTN Framework is licensed under MITpurl PURL: https://gxy.io/GTN:T00306version Revision: 8
Small Non-coding RNAs (ncRNAs) play a vital role in many cellular processes such as RNA splicing, translation, gene regulation. The small RNA-seq is a type of RNA-seq in which RNA fragments are size selected to capture only short RNAs. One of the most common applications of the small RNA-seq is discovering novel small ncRNAs. Mapping the small RNA-seq data reveals interesting patterns that represent the traces of the small RNA processing.
For example, consider the miRNA biogenesis. The primary miRNA transcripts are processed by Drosha-complexes and results in hairpin precursor miRNAs. Then after they transported to the cytoplasm, Dicer slices off the hairpin. One of the RNA strands bound by Argonaute proteins regulates the target mRNA while the other strand is degraded.
The following figure (bottom part of the figure is taken from Pasquinelli 2012) represents one of the small RNA processing patterns. The vertical block bars represent a mapped reads on reference genome. The height of the bars represents the number of reads mapped at that location. Reads in each bubble represent a read profile. In this case, they both are from miRNAs. From the small RNA-seq, we often see the two processed miRNA strands after mapping. The miRNA strand which targets the mRNA is expressed and we see more reads compared to the degraded strand. The gap between those two piles of reads represents the missing hairpin.
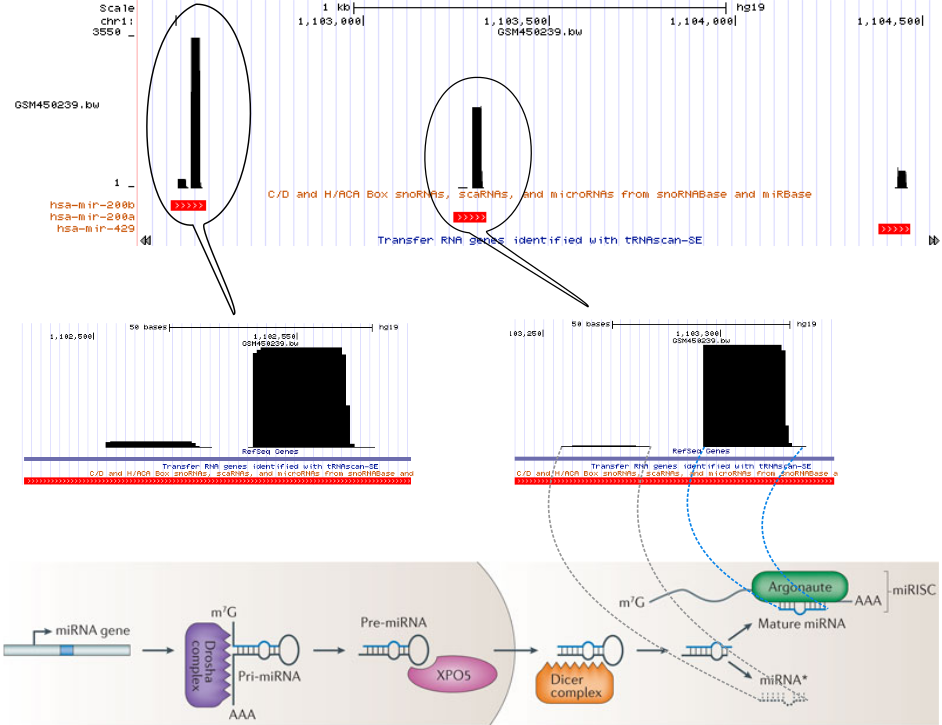 Open image in new tab
Open image in new tabIn this tutorial, we will learn how to use BlockClust to cluster similar processing patterns together.
AgendaIn this tutorial, we will cover:
Preprocessing of the data
The data is we use is from human cell line MCF-7 sequenced in single-end on Illumina Genome Analyzer II. Reads are 36nt long and should be sufficiently long enough for this type of data. We start with a BAM file as an input for this tutorial. This BAM file is a result of mapping the reads on human reference genome build hg19 using segemehl. If you want to use your own raw sequencing data, please clip the adapters and map to the reference genome.
In the first step, we convert the BAM file into BED file. This is not a plain file conversion. Hence we cannot use any kind of BAM to BED conversion tools. We use BlockClust tool in the pre-processing mode for this purpose. The resulting BED file contains tags (a tag is a unique read sequence in a deep-sequencing library) and their normalized expression (in column 5), i.e. the ratio of the read count per tag to the number of mappings on the reference genome.
Get data
Hands-on: Data upload
Create a new history for this tutorial
To create a new history simply click the new-history icon at the top of the history panel:
Import the files from Zenodo
https://zenodo.org/record/2172221/files/GSM769512.bam
- Copy the link location
Click galaxy-upload Upload Data at the top of the tool panel
- Select galaxy-wf-edit Paste/Fetch Data
Paste the link(s) into the text field
Press Start
- Close the window

QuestionWhy is it mandatory to clip the adapters from the small RNA-seq data?
Generally, the processed RNA fragments from the small RNAs are about 18-30nt long. If the sequenced read length is longer than the fragment (which is usually the case), the sequencer reads into the 3’ adapter.
Sort BAM file
Before continue to BAM to BED conversion we need to sort the alignments in the input BAM file by their positions.
Hands-on: Sort BAM
- Samtools sort tool with the following parameters:
- param-file “BAM File”:
GSM769512.bam
BAM to BED of tags
Now it is time to do the actual conversion.
Hands-on: BlockClust preprocessing
- BlockClust tool with the following parameters:
- “Select mode of operation”:
Pre-processing- param-file “BAM file containing alignments”: output of Samtools sort tool
Grouping of the adjacent reads into read profiles
Now we group the adjacent reads into so-called blocks and blockgroups using blockbuster tool. In general, each blockgroup should represent a single ncRNA read profile.
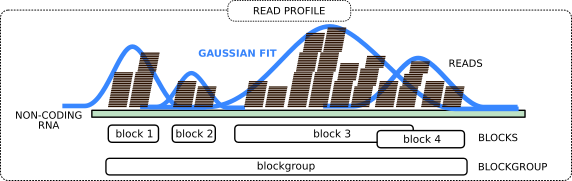 Open image in new tab
Open image in new tabThe idea is to perform peak detection on the signal obtained by counting the number of reads per nucleotide. This signal, spanning adjacent loci, is then modeled with a mixture of Gaussians. An iterative greedy procedure is then used to collect reads that belong to the same block, starting from the largest Gaussian component, and removing them in successive iterations. The tool further assembles a sequence of adjacent blocks into a blockgroup if the blocks are either overlapping or are at a distance smaller than a user-defined threshold. A more detailed explanation is at the blockbuster website
Sort the BED file of tags
In order to run the blockbuster successfully the input BED file need to be sorted by chromosome, strand, start position and then end positions.
Hands-on: Sort the BED file
- Sort tool with the following parameters:
- param-file “Sort Query”: output of BlockClust tool
- In “Column selections”:
- “on column”:
Column: 1- “Flavor”:
Alphabetical sort- Click on “Insert Column selections”:
- In “Column selections”:
- “on column”:
Column: 6- “Flavor”:
Alphabetical sort- Click on “Insert Column selections”:
- In “Column selections”:
- “on column”:
Column: 2- “Flavor”:
Fast numeric sort- Click on “Insert Column selections”:
- In “Column selections”:
- “on column”:
Column: 3- “Flavor”:
Fast numeric sort- “Ignore case”:
Yes
Group reads into blocks and blockgroups using blockbuster
Hands-on: blockbuster
- blockbuster tool with the following parameters:
- param-file “BED file containing read expressions”: output of Sort tool
- “Type of output”:
reads (blockbuster format)
Clustering with BlockClust
Here we use BlockClust in clustering mode. All you need here is the output of the blockbuster.
Apart from clustering, BlockClust has built-in class specific discriminative models for C/D box snoRNA, H/ACA box snoRNA, miRNA, rRNA, snRNA, tRNA and Y_RNA. So it can also be used to predict if a read profile might belong to one of the known ncRNA class.
Hands-on: clustering
- BlockClust tool with the following parameters:
- “Select mode of operation”:
Clustering and classification
- param-file “Input blockgroups file”: output of blockbuster tool
- “Select reference genome”:
Human (hg19)- “Would you like to perform classification?”:
Yes- “Mode of classification”:
Model based
The tool produces the following four output files.
The BlockClust: BED of predicted clusters file is the result of Markov cluster algorithm. Each entry in this file represents a blockgroup. The 4th column contains is in the form of annotation:blockgroup_id:cluster_id. The cluster_id represents which cluster the blockgroup belongs to.
The BlockClust: Model based predictions BED file is the result of classification. Each entry in this file represents a blockgroup. The 4th column contains the annotation of the ncRNA which it overlaps. For the blockgroups which are not overlapped with any known ncRNAs, the prefix predicted_ is added indicating that it is predicted by the BlockClust classification models.
The BlockClust: Hierarchical clustering plot file shows a dendrogram construced based on average linkage clustering.
The BlockClust: Pairwise similarities file contains the pairwise similarities of all input blockgroups.
Next, we will visualize the read profiles (from the BAM file) and the predictions (BED from BlockClust) together.
Hands-on: visualization
- Install IGV (if not already installed)
- Start IGV locally
- Expand the param-file
GSM769512.bamfile- Click on the
localindisplay with IGVto load the reads into the IGV browser- Expand the param-file
BlockClust: Model based predictions BEDfile- Click on the
localindisplay with IGVto load the reads into the IGV browser- Go to the location
chr4:90653059-90653141in IGV.
Question
- Do you see any already annotated transcript in that location?
- What does the read profile resemble?
- What did BlockClust predict?
- There is no annotated gene from RefSeq annotation on the same strand.
- It resembles the miRNA profiles mentioned in the introduction section. One of the read piles looks like mature miRNA and the other like miRNA*.
- Predicted as a miRNA.
CommentIn order for this step to work, you will need to have either IGV or Java web start installed on your machine. However, the questions in this section can also be answered by inspecting the IGV screenshots below.
Check the IGV documentation for more information.
Conclusion
In this tutorial, we learned how to use BlockClust to cluster real small RNA sequencing read profiles based on their similarity. We also learned that the BlockClust can also be used to classify the read profiles based on pre-built classification models.