Open your favorite browser (Chrome, Safari or Firefox as your browser, not Internet Explorer!)
Browse to your Galaxy instance
Log in or register
Comment: Different Galaxy servers
This is an image of Galaxy Australia, located at usegalaxy.org.au
The particular Galaxy server that you are using may look slightly different and have a different web address.
You can also find more possible Galaxy servers at the top of this tutorial in Available on these Galaxies
The Galaxy homepage is divided into four sections (panels):
The Activity Bar on the left: This is where you will navigate to the resources in Galaxy (Tools, Workflows, Histories etc.)
Currently active “Activity Panel” on the left: By default, the toolTools activity will be active and its panel will be expanded
Viewing panel in the middle: The main area for context for your analysis
History of analysis and files on the right: Shows your “current” history; i.e.: Where any new files for your analysis will be stored
The first time you use Galaxy, there will be no files in your history panel.
Key Galaxy actions
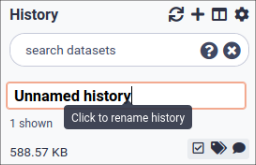
Name your current history
Your “History” is in the panel at the right.
Hands-on: Name history
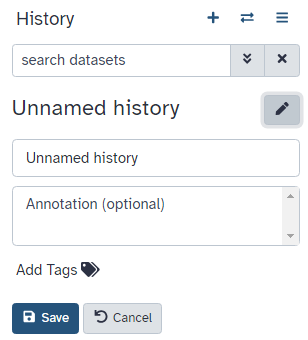
Go to the History panel (on the right)
Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
Comment
In some previous versions of Galaxy, you will need to click on the history name to rename it as shown here:
Type in a new name, for example, “My Analysis”
Click on Save
Comment: Renaming not an option?
If renaming does not work, it is possible you aren’t logged in, so try logging in to Galaxy first. Anonymous users are only permitted to have one history, and they cannot rename it.
Upload a file
The “Activity Bar” can be seen on the left-most part of the interface.
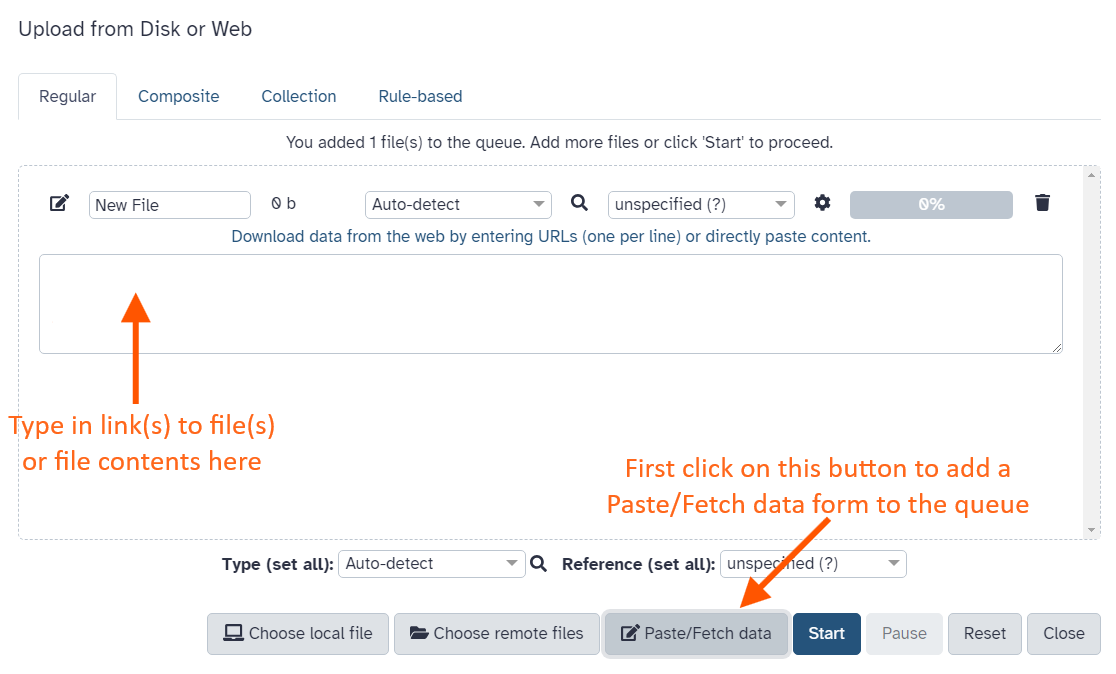
Hands-on: Upload a file from URL
At the top of the Activity Bar, click on the galaxy-uploadUpload activity
Your uploaded file is now in your current history.
When the file has uploaded to Galaxy, it will turn green.
Comment
After this you will see your first history item (called a “dataset”) in Galaxy’s right panel. It will go through
the gray (preparing/queued) and yellow (running) states to become green (success).
Sometimes during courses, data upload gets a little slow. You can also import data through a history link.
Figure 1: A FastQ file of course has four lines per record: the record identifier (`@mutant-no_snps.gff-24960/`), the sequence (`AATG…`), the plus character (`+`), and then the quality scores for the sequence (`5??A…`).
Use a tool
Let’s look at the quality of the reads in this file.
Hands-on: Use a tool
Type FastQC in the tools panel search box (top)
Click on the FastQC ( Galaxy version 0.74+galaxy1) tool
The tool will be displayed in the central Galaxy panel.
Select the following parameters:
param-file“Raw read data from your current history”: the FASTQ dataset that we uploaded
No change in the other parameters
Click Execute
This tool will run and two new output datasets will appear at the top of your history panel.
Tools are frequently updated to new versions. Your Galaxy may have multiple versions of the same tool available. By default, you will be shown the latest version of the tool. This may NOT be the same tool used in the tutorial you are accessing. Furthermore, if you use a newer tool in one step, and try using an older tool in the next step… this may fail! To ensure you use the same tool versions of a given tutorial, use the Tutorial mode feature.
Open your Galaxy server
Click on the curriculum icon on the top menu, this will open the GTN inside Galaxy.
Navigate to your tutorial
Tool names in tutorials will be blue buttons that open the correct tool for you
Note: this does not work for all tutorials (yet)
You can click anywhere in the grey-ed out area outside of the tutorial box to return back to the Galaxy analytical interface
Warning: Not all browsers work!
We’ve had some issues with Tutorial mode on Safari for Mac users.
Try a different browser if you aren’t seeing the button.
View results
We will now look at the output dataset called FastQC on data 1: Webpage.
Comment
Note that Galaxy has given this dataset a name according to both the tool name (“FastQC”) and the input (“data 1”) that it used.
The name “data 1” means the dataset number 1 in Galaxy’s current history (our FASTQ file).
Hands-on: View results
Once it’s green, click on the galaxy-eye (eye) icon next to the “Webpage” output dataset.
The information is displayed in the central panel
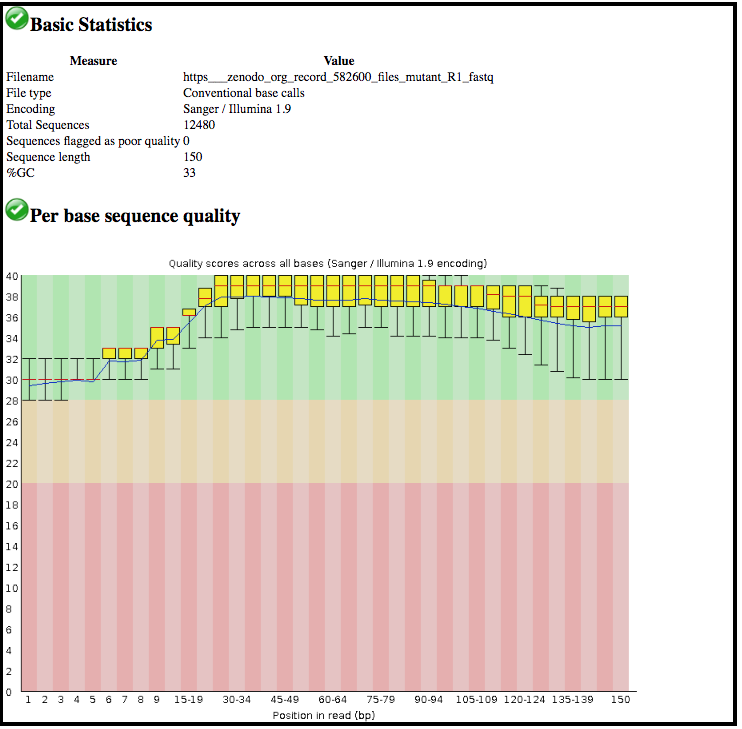
This tool has summarised information about all of the reads in our FASTQ file.
Question
What was the length of the reads in the input FASTQ file?
Do these reads have higher quality scores in the centre or at the ends?
150 bp
In the center
Run another tool
Let’s run a tool to filter out lower-quality reads from our FASTQ file.
Hands-on: Run another tool
Type Filter by quality in the tools panel search box (top)
Click on the tool Filter by quality ( Galaxy version 1.0.2+galaxy2)
“Percent of bases in sequence that must have quality equal to / higher than cut-off value”: 80
Click Execute
After the tool has run, its output dataset will appear at the top of your History panel.
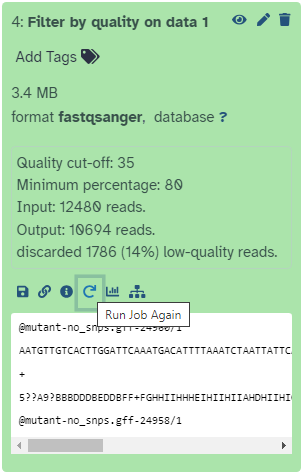
This dataset will be called “Filter by quality on data 1”.
Remember that Galaxy has named this file according to the tool it used (“Filter by quality”) and the input dataset (“data 1”).
The actual numbers in front of the datasets in the history are not important.
What are the results from this filtering tool?
We could click on the eye icon to view the contents of this output file, but it will not be very informative - we will just see a list of reads.
Hands-on: Get metadata about a file
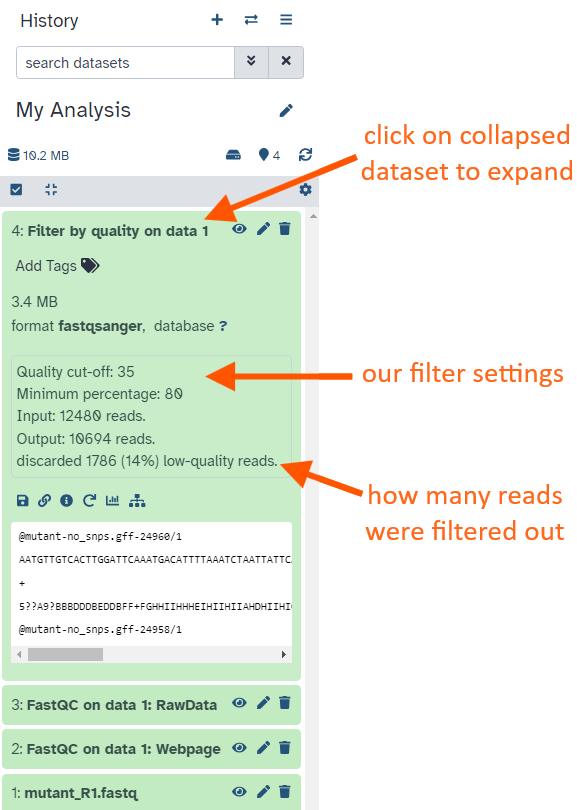
Click on the dataset (around where its name is) in the History panel.
This expands the information about the file. (By default all datasets are collapsed)
Question
How many reads have been discarded?
1786 low-quality reads were discarded
Re-run that tool with changed settings
We can now try to filter our input reads to an even higher standard, and see how this changes the resulting output (an exploratory analysis). We will change the filter settings and re-run the tool.
Hands-on: Re-run the tool
Click on the galaxy-refresh icon (Run Job Again) for the output dataset of Filter by qualitytool
This brings up the tool interface in the central panel with the parameters set to the values used previously to generate this dataset.
Change the settings to something even stricter
For example, you might decide you want 80 percent of bases to have a quality of 36 or higher, instead of 35.
Click Execute
View the results: Click on the output dataset name to expand the information
Comment
Not the galaxy-eye (eye) icon.
Question
How many reads were discarded under these new filtering conditions?
If you selected 80% of bases with 36 as quality cut-off, then 11517 reads (92%) should have been discarded, which indicates that we have gone too far with the filtering in this case.
You can re-run a tool many times with different settings. Each time you re-run the tool, its new output datasets will appear at the top of your current history.
Share your history
Finally, let’s imagine that you had a problem in your analysis and you want to ask for help. The easiest way to ask for help is to share your history. Try and create a link for your history and share it with…yourself!
Sharing your history allows others to import and access the datasets, parameters, and steps of your history.
Access the history sharing menu via the History Options dropdown (galaxy-history-options), and clicking “history-share Share or Publish”
Share via link
Open the History Optionsgalaxy-history-options menu at the top of your history panel and select “history-share Share or Publish”
galaxy-toggleMake History accessible
A Share Link will appear that you give to others
Anybody who has this link can view and copy your history
Publish your history
galaxy-toggleMake History publicly available in Published Histories
Anybody on this Galaxy server will see your history listed under the Published Histories tab opened via the galaxy-histories-activityHistories activity
Share only with another user.
Enter an email address for the user you want to share with in the Please specify user email input below Share History with Individual Users
Your history will be shared only with this user.
Finding histories others have shared with me
Click on the galaxy-histories-activityHistories activity in the activity bar on the left
Click the Shared with me tab
Here you will see all the histories others have shared with you directly
Note: If you want to make changes to your history without affecting the shared version, make a copy by going to History Optionsgalaxy-history-options icon in your history and clicking Copy this History
Convert your analysis history into a workflow
When you look carefully at your history, you can see that it contains all the steps of our analysis, from the beginning (at the bottom) to the end (on top). The history in Galaxy records details of every tool you run and preserves all parameter settings applied at each step. But when you need to analyze new data, it would be tedious to do each step one-by-one again. Wouldn’t it be nice to just convert this history into a workflow that we will be able to execute again and again?
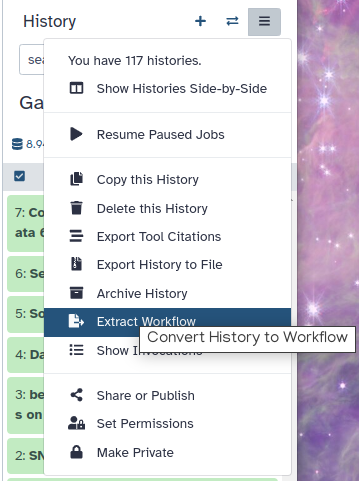
Galaxy makes this very easy with the Extract workflow option. This means any time you want to build a workflow, you can just perform the steps once manually, and then convert it to a workflow, so that next time it will be a lot less work to do the same analysis.
Hands-on: Extract workflow
Clean up your history: remove any failed (red) jobs from your history by clicking on the galaxy-delete button.
This will make the creation of the workflow easier.
Click on galaxy-history-options (History options) at the top of your history panel and select Extract workflow.
The central panel will show the content of the history in reverse order (oldest on top), and you will be able to choose which steps to include in the workflow.
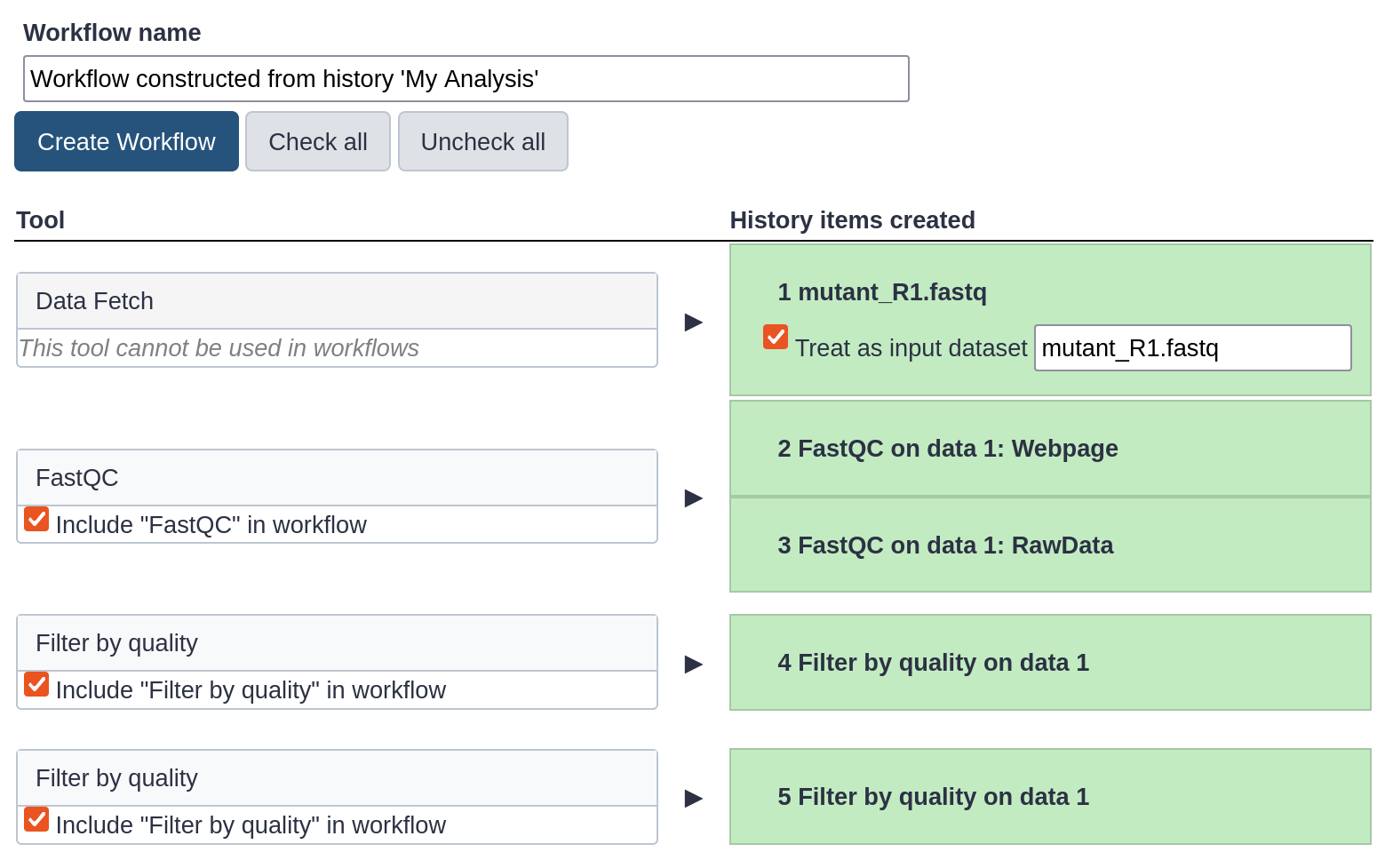
Replace the Workflow name to something more descriptive, for example: QC and filtering.
Rename the workflow input in the box at the top of second column to: FASTQ reads
If there are any steps that shouldn’t be included in the workflow, you can uncheck them in the first column of boxes. In this case, uncheck the second Filter by quality tool at the bottom, where we used a too high quality cut-off.
Click on the Create Workflow button near the top.
You will get a message that the workflow was created.
In a minute we will see how to find the extracted workflow and how to use it.
Create a new history
Let’s create a new history.
Hands-on: New history
Create a new history
To create a new history simply click the new-history icon at the top of the history panel:
Rename your history, e.g. “Next Analysis”
Click on galaxy-pencil (Edit) next to the history name (which by default is “Unnamed history”)
Type the new name
Click on Save
To cancel renaming, click the galaxy-undo “Cancel” button
If you do not have the galaxy-pencil (Edit) next to the history name (which can be the case if you are using an older version of Galaxy) do the following:
Click on Unnamed history (or the current name of the history) (Click to rename history) at the top of your history panel
Type the new name
Press Enter
This new history does not have any datasets in it yet.
Look at multiple histories
Where is your first history, called “My Analysis”?
Hands-on: View histories in History Multiview
One of the ways to view multiple histories at once in Galaxy is through the History Multiview
There are multiple ways to get to the multview:
Click on the galaxy-multihistoryHistory Multiview activity in the activity bar
Or, click on galaxy-history-options (History options) and then click on the galaxy-columnsShow Histories side-by-side option
A new page will appear with your histories displayed side-by-side here.
Copy a dataset into your new history
Click on the FASTQ dataset in “My Analysis” history
Figure 2: Copy a dataset between histories by dragging it
This makes a copy of the dataset in the new history (without actually using additional disk space).
Click on the Home icon galaxy-home (or Analyze Data on older versions of Galaxy) in the top panel to go back to your analysis window
Your main Galaxy window will now show “Next Analysis” as the current history, and it will have one dataset in it.
Comment
This is not the only way to view your histories in Galaxy:
An exhaustive list (table) of all your histories is available in the My Histories tab in the Histories List accessible via clicking the galaxy-histories-activityHistories activity
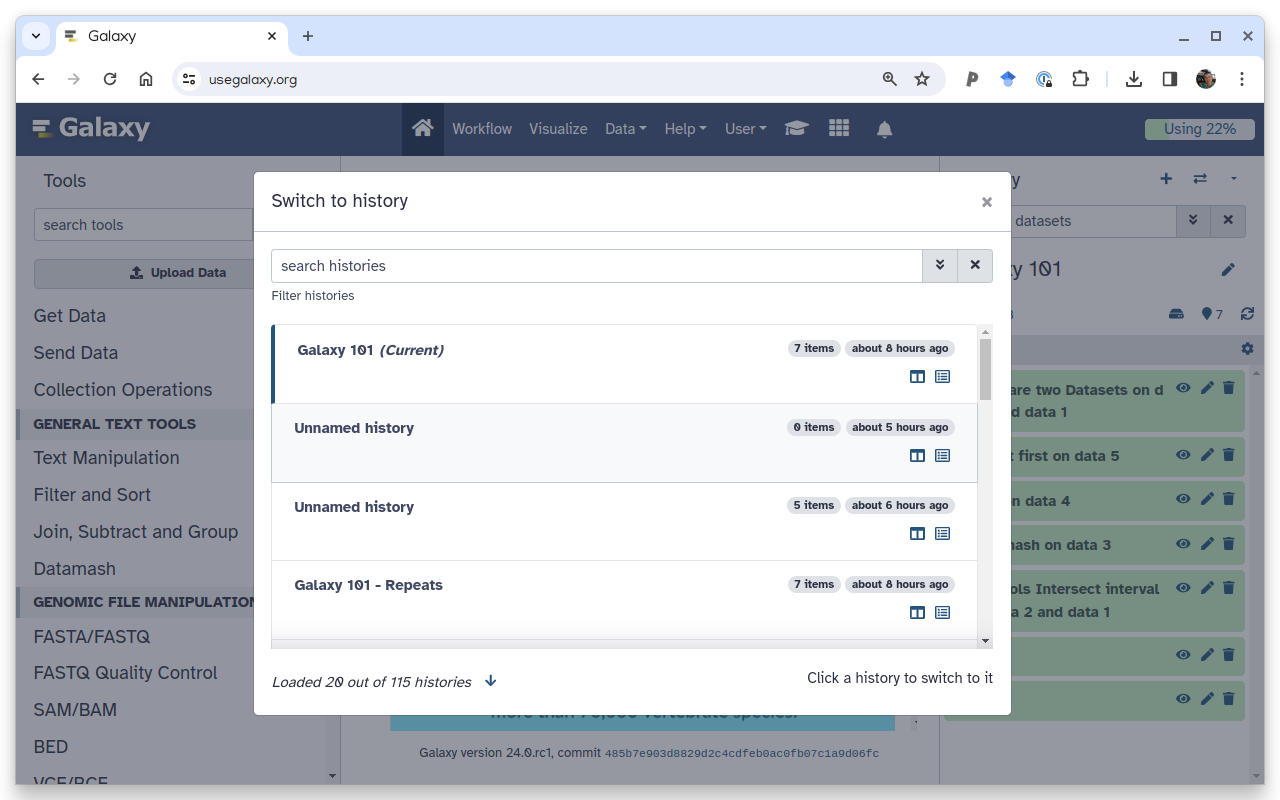
You can quickly switch to another history by clicking on the switch-historiesSwitch to history button next to galaxy-history-optionsHistory options:
To switch to an existing history simply click the switch-histories icon at the top of the history panel. This opens a list of histories existing in a given Galaxy account in the middle part of the interface.
Now that we have built our workflow, let’s use it to re-create our small analysis in a single step. The same workflow could also be used on some new FASTQ data to quickly repeat the same analysis on different inputs.
Hands-on: Run workflow
Click on the galaxy-workflows-activityWorkflows activity in the activity bar.
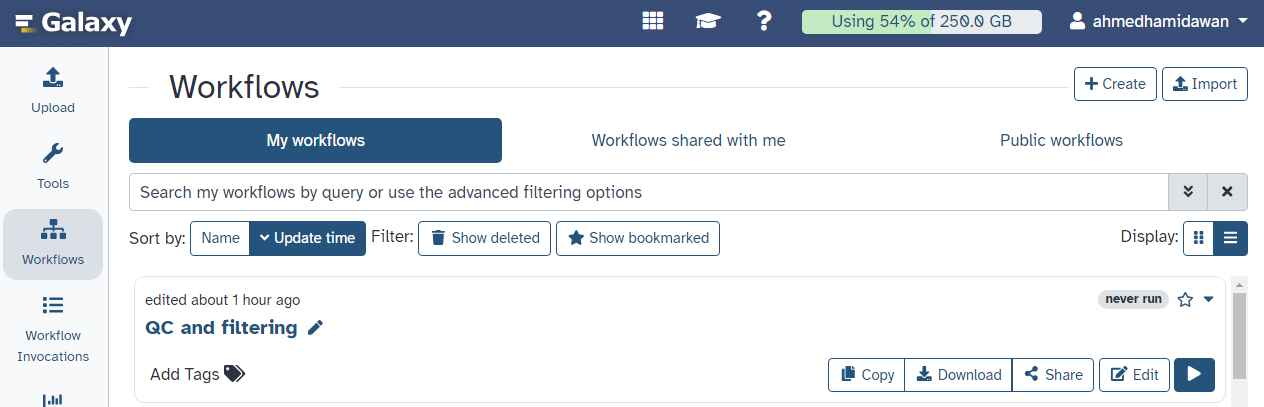
Here you have a list of all your workflows (the My Workflows tab is active by default).
Your newly created workflow should be listed at the top:
You can see all available actions for the workflow on the workflow card, e.g. edit, copy, rename, share etc. Any other options (e.g.: delete, export etc.) are available by clicking on the galaxy-dropdownWorkflow actions button on the top right of the card.
Click on the workflow-run (Run workflow) button on the bottom right of the workflow card.
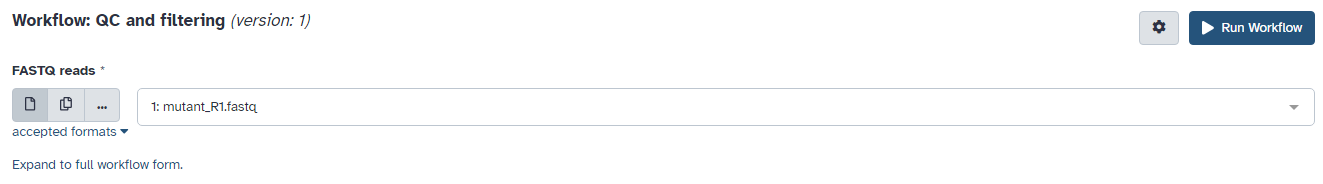
The central panel will change to allow you to configure and launch the workflow.
Check that the “FASTQ reads” input is set to the FASTQ dataset we have copied to the new history.
In this page we could change any parameter for the tools composing the workflow as we would do when running them one by one.
Click the Run Workflow button at the top-right of the screen.
You should see a message that the workflow was successfully invoked. Then jobs will start to run and datasets appear in your “Next Analysis” history, replicating the steps of your previous history.
Conclusion
Well done! You have completed the short introduction to Galaxy, where you named the history, uploaded a file, used a tool, viewed results and run a workflow. Additional tutorials are available for a more in-depth introduction to Galaxy’s features.
You've Finished the Tutorial
Please also consider filling out the Feedback Form as well!
Key points
The Galaxy interface has an activity bar on the left, a tool (or other activated) panel next to it (if expanded), viewing pane in the middle, and a history of your data analysis on the right.
You can create a new history for each analysis. All your histories are saved.
To get data into Galaxy, you can upload a file by pasting in a web address. There are other ways to get data into Galaxy (not covered in this tutorial): you can upload a file from your computer, and you can import an entire history.
Choose a tool and change any settings for your analysis.
Run the tool. The output files will be saved at the top of your history.
View the output files by clicking on the eye icon.
View all your histories and move files between them. Switch to a different history.
Log out of your Galaxy server. When you log back in (to the same server), your histories will all be there.
Did you use this material as an instructor? Feel free to give us feedback on how it went.
Did you use this material as a learner or student? Click the form below to leave feedback.
Hiltemann, Saskia, Rasche, Helena et al., 2023 Galaxy Training: A Powerful Framework for Teaching! PLOS Computational Biology 10.1371/journal.pcbi.1010752
Batut et al., 2018 Community-Driven Data Analysis Training for Biology Cell Systems 10.1016/j.cels.2018.05.012
@misc{introduction-galaxy-intro-short,
author = "Anna Syme and Nicola Soranzo",
title = "A short introduction to Galaxy (Galaxy Training Materials)",
year = "",
month = "",
day = ""
url = "\url{https://training.galaxyproject.org/training-material/topics/introduction/tutorials/galaxy-intro-short/tutorial.html}",
note = "[Online; accessed TODAY]"
}
@article{Hiltemann_2023,
doi = {10.1371/journal.pcbi.1010752},
url = {https://doi.org/10.1371%2Fjournal.pcbi.1010752},
year = 2023,
month = {jan},
publisher = {Public Library of Science ({PLoS})},
volume = {19},
number = {1},
pages = {e1010752},
author = {Saskia Hiltemann and Helena Rasche and Simon Gladman and Hans-Rudolf Hotz and Delphine Larivi{\`{e}}re and Daniel Blankenberg and Pratik D. Jagtap and Thomas Wollmann and Anthony Bretaudeau and Nadia Gou{\'{e}} and Timothy J. Griffin and Coline Royaux and Yvan Le Bras and Subina Mehta and Anna Syme and Frederik Coppens and Bert Droesbeke and Nicola Soranzo and Wendi Bacon and Fotis Psomopoulos and Crist{\'{o}}bal Gallardo-Alba and John Davis and Melanie Christine Föll and Matthias Fahrner and Maria A. Doyle and Beatriz Serrano-Solano and Anne Claire Fouilloux and Peter van Heusden and Wolfgang Maier and Dave Clements and Florian Heyl and Björn Grüning and B{\'{e}}r{\'{e}}nice Batut and},
editor = {Francis Ouellette},
title = {Galaxy Training: A powerful framework for teaching!},
journal = {PLoS Comput Biol} Computational Biology}
}
Funding
These individuals or organisations provided funding support for the development of this resource
5 stars:
Liked: All topics under this session
Disliked: Nothing! All points adressed are very informative
5 stars:
Liked: The images, organization and teaching.
Disliked: For me, it's already great. I was able to clearly understand the content.
5 stars:
Liked: very clear and easy to follow
5 stars:
Liked: No topic was left without a proper closure. Everything was explained to the utmost detail and considering common pitfalls user journeys may have.
Disliked: I'd like to have the chance to answer the questions before watching the right responses. That way, I will be able to actively check whether or not I'm understanding not only the tools but also the concepts and tasks behind them.
October 2024
5 stars:
Liked: Very clear and up to date links and screen captures
5 stars:
Liked: Easy to play
Disliked: Add different languages
5 stars:
Liked: level of detail and thorough instructions
4 stars:
Liked: Practical exercizes in parallel with explanaition
Disliked: Maybe having this step by step exercizes inside the Galaxy web page
5 stars:
Liked: simple explanation to the website
5 stars:
Liked: The interface was galaxy is very friendly
Disliked: nil
5 stars:
Liked: It was short and precise.
5 stars:
Liked: Precise work description
Disliked: Maybe upload some datasets for people learning from beginning, but I found them at different tutorial (https://training.galaxyproject.org/training-material/topics/galaxy-interface/tutorials/collections/tutorial.html#tip-upload-fastqsanger-datasets-via-links)
5 stars:
Liked: Step by step instructions that are easy to follow!!
5 stars:
Liked: Simplicity and easy to follow diagrams
5 stars:
Liked: Nicely represented
September 2024
5 stars:
Liked: Very clear and easy to follow
5 stars:
Liked: Clear and simple.
Disliked: I would like to learn where to look for details regarding the function of each tool.
5 stars:
Liked: all of it
5 stars:
Liked: I enjoyed the layout overall with the helpful visuals added to make sure you are completing the correct action, overall great training!
5 stars:
Liked: Everything was so clear and easy to understand
5 stars:
Liked: Really fast and nicely made
5 stars:
Liked: everything, however i need to say it was challenging.
Disliked: need a bit more time for activities, for people who struggling with it tools.
5 stars:
Liked: Introduction to Galaxy analysis software.
Disliked: N/A.
2 stars:
Liked: That there were screenshots that visually aided the steps and allowed for better comprehension of the method steps.
Disliked: More precise language within steps would be nice. For instance instead of saying copy a file, saying to copy the link of the file below when uploading would be more accurate as the person would then be able to understand to copy THAT file, instead of wandering what file to upload. Additionally, the other word to improve would be to replace the word "execute" with "run tool" because there is no "execute" button to run the tool, only the "run tool" button which does the same thing.
5 stars:
Liked: I managed to do all the activities, I feel I can do.
Disliked: Personally, the opportunity to continue from the basic to the advanced.
5 stars:
Liked: Picture window
Disliked: More pictures and motion graphics
5 stars:
Liked: Easy to follow
4 stars:
Liked: User friendly, informative
Disliked: Smaller screen graphics so it is easily to view entire set of instructions for each step.
4 stars:
Liked: A good level for someone who is not comfortable with using software packages
Disliked: A summary of key buttons used in the tutorial for quick reference that can be printed would be useful
5 stars:
Liked: proper instructions, easy to follow, detailed explannation
5 stars:
Liked: The layout and the defined tutorial were very understanding
5 stars:
Liked: The style of your tutorial
August 2024
5 stars:
Liked: It was easy to follow.
5 stars:
Liked: Ive used this server before, but strangely a year on, I understand it much better.
Disliked: some of the tabs, are now different, updating these on this tutorial would make it easier, but by trial and error easy to figure out.
5 stars:
Liked: As a Bioinformatician I like this website to input the Data in a upload file.
Disliked: I improve new thinks by read this tutorial.
5 stars:
Disliked: I didn't like how you have to swap tabs so much.
5 stars:
Liked: lo interactivo de sus tutoriales
5 stars:
Liked: It's complete and easy to understand.
5 stars:
Liked: it was hands on and i learned a lot.
5 stars:
Liked: easy
5 stars:
Liked: How I got to use the tools myself and mess around with the parameters if I wanted
5 stars:
Liked: Very understandable and easy to follow
Disliked: everything was great
3 stars:
Liked: logical and clear structure
5 stars:
Liked: easy to follow
July 2024
5 stars:
Liked: clear and straight forward instructions
4 stars:
Liked: You learn fast essential concepts
5 stars:
Liked: I liked the fact that the tutorial was very detailed
Disliked: I don't know the purpose of the timer attached to the first slide.
5 stars:
Liked: It was clear and concise
June 2024
5 stars:
Liked: every thing
Disliked: videos
5 stars:
Liked: I liked the examples and the tips it gave. It also had interactive questions to help us navigate and make sure we were finding everything.
Disliked: I think it'd be good to example some aspects of the software. For example, what is this function's purpose and why do we use it?
4 stars:
Liked: Detailed steps and explanation
5 stars:
Liked: Training content, the flow of information
5 stars:
Liked: clear instructions
5 stars:
Liked: Clear instruction
Disliked: Nothing really from my end
5 stars:
Liked: simple and straight forward tutorial
Disliked: I don't know currently.
5 stars:
Liked: Run workflow in the new history, Re-run that tool with changed settings
Disliked: It was a bit tricky to find FastQC ( Galaxy version 0.73+galaxy0)
5 stars:
Liked: I like the simplicity, straightforward and professional crafting of the material. It was extraordinarily easy to follow. I sincerely appreciate the team's effort and generosity in preparing a training of this nature. I find it a great pleasure to have been accepted for the training.
Disliked: I'm fully satisfied with the material and structure of the training thus far. Perhaps I may identify areas that could need improvement in subsequent tutorials.
5 stars:
Liked: user friendly, easy to follow
Disliked: don't know
5 stars:
Liked: The simplicity of the interface
Disliked: NAP
5 stars:
Liked: The descriptions of the steps and instructions are very clear
5 stars:
Liked: Easy to understand and to follow
Disliked: more screenshots would be better but in general it was great
5 stars:
Liked: It was self explaining and i could follow easily without a physical instructor
Disliked: Some of the tabs naming have changed, this could be updated in the manual
4 stars:
Disliked: My interface was slightly different from that assumed/shown in the tutorial. I didn't see a link to a 'next' tutorial.
4 stars:
Liked: Easy to follow. Expandable bits for things I should remember but maybe don't.
Disliked: My interface was slightly different from that assumed/shown in the tutorial.
4 stars:
Liked: good examples and demos
Disliked: more examples and demos
May 2024
5 stars:
Liked: easy to do
Disliked: none
3 stars:
Disliked: there is a lot of the botto,s that is not here
5 stars:
Liked: well described
5 stars:
Liked: I like it a lot.
Disliked: As I am a novice in bioinformatics, I find silly question for skilled person hard for me. such as, why we are adding cut off value 35 or How to use that out put in research? thank you so much.
5 stars:
Liked: Step by step detail guide
Disliked: Short video as well would be nice
5 stars:
Liked: The steps were easy to follow, especially with the example images.
Disliked: Provide more updated example images for the second analysis.
April 2024
5 stars:
Liked: Simplicity
Disliked: new versions
5 stars:
Liked: Easy to understand even for beginners
Disliked: maybe you can add how other type of data to "upload data"
5 stars:
Liked: All content
2 stars:
Liked: difficult to follow
Disliked: to detail each step
5 stars:
Liked: The walkthrough was so distributive and easy to comprehend
Disliked: Nothing that I can think of can be improved for this week's materials
March 2024
5 stars:
Liked: Explication avec des exemples très efficace !
5 stars:
Liked: Helpfull
Disliked: more vidios
5 stars:
Liked: Simple and easy to follow. Helpful to be hands on with the data set.
Disliked: Nothing
5 stars:
Liked: It was really good, very clear and helpfull!
Disliked: n.a.
5 stars:
Liked: Really easy and descriptive language
Disliked: maybe include some short videos as the one of copying the fasta to another histry
5 stars:
Liked: Very well structured and very detailed information on every step.
Disliked: Maybe other options of how to upload files could be explained
February 2024
5 stars:
Liked: Well guided and great information
5 stars:
Liked: Everything
Disliked: All good
5 stars:
Liked: Clear and easy to follow guide
Disliked: A little figure schematic pointing out where each thing is rather tyhen just descriptive text
5 stars:
Liked: los ejemplos con imágenes
Disliked: tal vez poner ejemplos de otras herramientas
5 stars:
Liked: step for step explanation supported by fotos and videos, easy language
4 stars:
Liked: Detailed step by stepintroduction into the application
Disliked: It seems to me like the tutorial was written for an earlier version of Galaxy. In the beginning some buttons were named differently from the current panels, which was a bit confusing at first.
5 stars:
Liked: Clear instructions. Liked the image and drop down menu's.
5 stars:
Liked: The structure.
January 2024
5 stars:
Liked: simple to be understand
5 stars:
Disliked: really great session
5 stars:
Liked: everything worked as described
Disliked: double check that terms "run" and "execute" match up between tutorial and latest software
December 2023
4 stars:
Liked: It was clear
5 stars:
Liked: The screenshots/videos were helpful
4 stars:
Liked: easy to follow
Disliked: please add more screenshots for easy localization of the buttons
Questions:



 Open image in new tab
Open image in new tab

Open image in new tab